包括光固化粘合剂和热发泡剂的可再剥离粘合剂及包括其的可再剥离胶带的制作方法
1.本发明涉及包括光固化粘合剂和热发泡剂的可再剥离粘合剂及包括其的可再剥离胶带,更加具体地,所述粘合剂保持优秀的粘合力,且经紫外线照射和加热后易于从被粘物脱离。
背景技术:
2.丙烯酸发泡体型胶带(在基材及/或者粘合剂层中含微粒的丙烯酸类胶带)作为具有含微粒的压敏粘合剂层的压敏胶带,需要常温下的粘合强度或者抗剪强度等,例如常常用于粘合汽车、机械零件、电子零件、建材等各种领域的零件。
3.在这种状况下,现有的丙烯酸发泡体型胶带通过高粘合强度而具有高粘合可靠性,但由于其高粘合强度,反倒很难剥离、拆卸粘合部。
4.作为这种已知的丙烯酸发泡体型胶带,公开了在用于形成粘合剂层或者丙烯酸类粘合剂层的组合物中分散有玻璃微泡(中空微球)的压敏胶带(参照日本专利公告(昭)57-17030号及日本专利公开(平)7-48549号)。虽然这种发泡体型胶带显示非常高的剥离强度,但剥离时,由于其高剥离强度而不容易剥离。
5.对此,本发明人在为了解决上述问题而研究的过程中,发现了当通过使丙烯酸共聚物、光固化丙烯酸酯单体等具有规定组成的组合物聚合形成光固化粘合剂并以该光固化粘合剂和热发泡剂制备可再剥离粘合剂时,保持优秀的粘合力,且经紫外线照射和加热后,能够完全从被粘贴物脱离,而没有粘合剂转移,由此完成本发明。
技术实现要素:
6.技术问题
7.本发明为了解决上述现有技术的问题而提出,其目的在于,提供一种可再剥离粘合剂,其包括光固化粘合剂组合物及热发泡剂。
8.另外,其目的在于,提供一种可再剥离胶带,其通过将所述粘合剂涂布于基材而制备。
9.即,现有的发泡胶带通过加热来易于降低粘合力,但在被粘物上可能细微地残留粘合剂或者发泡剂残留物,并且,光固化胶带能够通过紫外线(uv)照射来消除几乎全部粘合力,但由于被使用的基材的湿润性,不容易脱离。
10.另外,表面的平滑度和湿润性优秀的被粘物及用于加压工艺中的被粘物,在工艺中持续增加粘合力,从而加工后相对难以脱离。因此,本发明的目的在于,提供一种可再剥离粘合剂及包括其的可再剥离胶带,该可再剥离粘合剂通过第一次紫外线照射进行光固化后,通过第二次加热进行发泡,从而能够以物理、化学方法完全剥离,而不会在被粘物上转移粘合剂或产生发泡剂的残留物。
11.技术方案
12.作为用于解决上述技术问题的技术方案,本发明的一实施方式提供一种可再剥离粘合剂,包括:光固化粘合剂,通过使包括丙烯酸共聚物、光固化丙烯酸酯类单体、光固化聚氨酯丙烯酸酯类低聚物、交联剂及光引发剂的光固化粘合剂组合物聚合而成;以及热发泡剂。
13.所述丙烯酸共聚物可以由碳原子数2至15的丙烯酸酯类单体聚合而成。
14.所述丙烯酸共聚物可以由选自丙烯酸-2-乙基己酯(2-ethylhexyl acrylate,2-eha)、乙酸乙烯酯(vinyl acetate,va)、甲基丙烯酸甲酯(methyl methacrylate,mma)、丙烯酸(acrylic acid,aa)及它们的组合中的单体聚合而成。
15.所述光固化丙烯酸酯类单体可以是包括选自1,4-己二醇二丙烯酸酯、1,6-丁二醇二丙烯酸酯、聚乙二醇二丙烯酸酯、季戊四醇三丙烯酸酯、三羟甲基丙烷三丙烯酸酯、双三羟甲基丙烷四丙烯酸酯、二季戊四醇五丙烯酸酯、季戊四醇五丙烯酸酯、季戊四醇四丙烯酸酯、三(2-羟烷基)异氰脲酸酯三环氧化物、三烯丙基异氰脲酸酯、三(2-羟烷基)异氰脲酸三丙烯酸酯、异佛尔酮二异氰脲酸酯、烷基二异氰脲酸酯、脂环族二异氰脲酸酯化合物、烷基二异氰脲酸酯三聚体、丙烯酸癸酯、丙烯酸月桂酯、丙烯酸硬脂酯、丙烯酸四氢糠酯、环三羟甲基甲缩醛丙烯酸酯、苯氧基聚乙二醇丙烯酸酯、丙烯酸苄酯、丙烯酸环氧乙酯、丙烯酸苯氧乙酯、三丙二醇二丙烯酸酯及它们的组合中的单体。
16.所述光固化聚氨酯丙烯酸酯类低聚物可以是包括具有2至6个官能团的脂肪族(aliphatic)聚氨酯丙烯酸酯。
17.所述交联剂可以是包括选自2,4-甲苯二异氰酸酯、2,6-甲苯二异氰酸酯、1,5-萘二异氰酸酯、二苯基甲烷二异氰酸酯、六亚甲基二异氰酸酯、异佛尔酮二异氰酸酯、对苯二甲基二异氰酸酯、间苯二甲基二异氰酸酯、二环己基甲烷二异氰酸酯、脂肪族多异氰酸酯及它们的组合中的交联剂。
18.所述光引发剂可以是包括选自二苯甲酮、苄基二甲基缩酮、安息香、安息香甲醚、安息香异丙醚、安息香异丁醚、安息香苯甲酸甲酯、安息香苯甲酸、苯乙酮、2,2-二甲氧基-2-苯基苯乙酮、2,4-二乙基噻唑酮、α-羟基环己基苯基酮、苄基二苯硫醚及它们的组合中的物质。
19.相对于100重量份的所述丙烯酸共聚物,所述光固化丙烯酸酯类单体的含量可以是1重量份至50重量份,所述光固化聚氨酯丙烯酸酯类低聚物的含量可以是5重量份至100重量份,所述交联剂的含量可以是0.1重量份至10重量份,所述光引发剂的含量可以是0.1重量份至10重量份。
20.所述热发泡剂可以是包括选自被微囊化的热膨胀微球、无机发泡剂、有机发泡剂及它们的组合中的物质。
21.相对于100重量份的所述光固化粘合剂,所述热发泡剂的含量可以是0.5重量份至50重量份。
22.将所述可再剥离粘合剂涂布于基材后,照射具有200nm至500nm波长的紫外线时,通过下述数学式1计算出的粘合力减少率为90%以上,
23.数学式1:
24.25.另外,根据本发明的另一实施方式,提供一种可再剥离胶带,通过将所述可再剥离粘合剂涂布于基材而制备。
26.所述可再剥离胶带可通过紫外线(uv)照射及加热来同时进行固化和发泡。
27.所述加热的温度可以为100℃至270℃。
28.发明效果
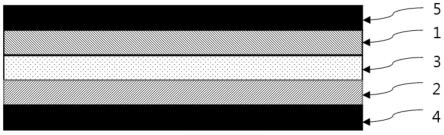
29.上述的本发明的包括光固化粘合剂及热发泡剂的可再剥离粘合剂对被粘物的湿润性优秀,从而具有高粘合力,且由于经紫外线(uv)照射和加热后,粘合力完全消失,更容易从被粘物脱离,并且在剥离时不会产生诸如粘合剂或者发泡粒子等残留物的污染,从而能够具有非常优秀的剥离性能。
附图说明
30.图1是示出本发明的一实施例的包括光固化粘合剂及热发泡剂的可再剥离胶带的使用状态的概略图。
31.附图标记:
32.1:第一粘合层
33.2:第二粘合层
34.3:基材层
35.4:第一剥离性薄膜层
36.5:第二剥离性薄膜层
具体实施方式
37.以下,更加详细说明本发明。但是本发明可以以各种不同形态实施,且本发明并非限制于此处说明的实施例,本发明仅由所附的权利要求书来定义。
38.另外,本发明中使用的术语只是为了说明特定的实施例而使用,并非旨在限定本发明。除非上下文另有明确规定,否则单数的表达包括复数的表达。在本说明书全文中,除非有相反的记载,否则“包括”某一构成要素是指可以进一步包括另一构成要素,而并非排除另一构成要素。
39.在对本发明进行具体描述以前,本发明的包括光固化粘合剂及热发泡剂的可再剥离粘合剂可以用作两面胶带,该两面胶带在需要对半导体、电气电子设备及移动设备等中所使用的模块部件、红外滤光片、玻璃、镜片等进行切割、研磨的工艺中,用于固定被粘物。即,在对所述被粘物进行切割或者研磨的过程中,夹具与被粘物之间不应发生脱离,因此用于固定它们的粘合剂需要强粘合力,并且需要作业后易于脱离的特性。对此,通常使用紫外光固化粘合剂,但常规使用的紫外光固化粘合剂存在被挤压的夹具和被粘物难以分离的缺点。另外,发泡胶带无法实现高粘合力,因此,在裁切和研磨时,被粘物不能良好地固定在夹具上,而易于分离,从而构成不良的原因。对此,本发明提出可再剥离粘合剂,其具有优秀的粘合力,易于调节初期粘合力(tack),且经紫外线(uv)固化和加热后完全消除粘合力,从而易于从夹具和被粘物中脱离。具体地,所述粘合剂包括光固化粘合剂及热发泡剂,通过第一次紫外线固化来消除粘合力,并进行第二次加热而使发泡剂膨胀,从而减少夹具和被粘物之间的表皮接触面积,从而易于脱离(分离)。
40.本发明的第一实施方式提供一种可再剥离粘合剂,其包括:光固化粘合剂,通过使包括丙烯酸共聚物、光固化丙烯酸酯类单体、光固化聚氨酯丙烯酸酯类低聚物、交联剂及光引发剂的光固化粘合剂组合物聚合而成;以及热发泡剂。
41.以下,参照图1对本发明的第一实施方式的可再剥离粘合剂进行详细说明。其中,所述图1是示出本发明的一实施例的可再剥离粘合剂的使用状态的概略图。
42.本发明的一实施例中,所述光固化粘合剂组合物可以包含丙烯酸共聚物。其中,所述丙烯酸共聚物由碳原子数2至15的丙烯酸酯类单体聚合而成,优选地,可以由选自丙烯酸-2-乙基己酯(2-ethylhexyl acrylate,2-eha)、乙酸乙烯酯(vinyl acetate,va)、甲基丙烯酸甲酯(methyl methacrylate,mma)、丙烯酸(acrylic acid,aa)及它们的组合中的单体聚合而成。另一方面,根据本发明的一实施例,所述丙烯酸共聚物可以由选自丙烯酸-2-乙基己酯(2-ethylhexyl acrylate,2-eha)、乙酸乙烯酯(vinyl acetate,va)、甲基丙烯酸甲酯(methyl methacrylate,mma)及丙烯酸(acrylic acid,aa)聚合而制备。
43.其中,相对于100重量份的所述丙烯酸-2-乙基己酯(2-ethylhexyl acrylate,2-eha),所述乙酸乙烯酯(vinyl acetate,va)的含量可以是20重量份至40重量份,所述甲基丙烯酸甲酯(methyl methacrylate,mma)的含量可以是10重量份至20重量份,所述丙烯酸(acrylic acid,aa)的含量可以是1重量份至20重量份。更加优选地,所述乙酸乙烯酯(vinyl acetate,va)的含量可以是29重量份至32重量份,所述甲基丙烯酸甲酯(methyl methacrylate,mma)的含量可以是14重量份至16重量份,所述丙烯酸(acrylic acid,aa)的含量可以是2重量份至10重量份。当单体的含量脱离所述范围时,由此制备的光固化粘合剂的粘合性能可能降低,且光固化后粘合力的消除程度可能微小。
44.本发明的一实施例中,所述丙烯酸共聚物可以是在溶剂及引发剂下,使所述丙烯酸酯类单体进行溶液聚合而制备的。其中,所述溶剂可以使用通常用于溶液聚合的溶剂,优选地,可以使用乙酸乙酯(ethyl acetate,ea)。另外,所述引发剂可以使用偶氮类引发剂,例如,可以使用2,2-偶氮二异丁腈(2,2-azobisisobutyronitrile,aibn)。另一方面,相对于100重量份的所述丙烯酸酯类单体,所述溶剂的含量可以是100重量份至200重量份,优选为130重量份至170重量份,根据本发明的一实施例可以约为150重量份。另外,相对于100重量份的所述丙烯酸酯类单体,所述引发剂的含量可以是0.001重量份至5重量份,优选为0.1重量份至2重量份,根据本发明的一实施例可以约为0.3重量份。
45.根据本发明的一实施例,所述光固化粘合剂组合物可以包含光固化丙烯酸酯类单体。其中,所述光固化丙烯酸酯类单体可以使用乙烯基为2至6个的丙烯酸单体,例如,所述光固化丙烯酸酯类单体可以是包括选自1,4-己二醇二丙烯酸酯、1,6-丁二醇二丙烯酸酯、聚乙二醇二丙烯酸酯、季戊四醇三丙烯酸酯、三羟甲基丙烷三丙烯酸酯、双三羟甲基丙烷四丙烯酸酯、二季戊四醇五丙烯酸酯、季戊四醇五丙烯酸酯、季戊四醇四丙烯酸酯、三(2-羟烷基)异氰脲酸酯三环氧化物、三烯丙基异氰脲酸酯、三(2-羟烷基)异氰脲酸三丙烯酸酯、异佛尔酮二异氰脲酸酯、烷基二异氰脲酸酯、脂环族二异氰脲酸酯化合物、烷基二异氰脲酸酯三聚体、丙烯酸癸酯、丙烯酸月桂酯、丙烯酸硬脂酯、丙烯酸四氢糠酯、环三羟甲基甲缩醛丙烯酸酯、苯氧基聚乙二醇丙烯酸酯、丙烯酸苄酯、丙烯酸环氧乙酯、丙烯酸苯氧乙酯、三丙二醇二丙烯酸酯及它们的组合中的单体。另一方面,根据本发明的一实施例,所述光固化丙烯酸酯类单体可以使用1,6-己二醇二丙烯酸酯(1,6-hexanediol diacrylate)。
46.在本发明的一实施例中,相对于100重量份的所述丙烯酸共聚物,所述光固化丙烯酸酯类单体的含量可以是1重量份至50重量份,优选为3重量份至20重量份,根据本发明的一实施例可以约为10重量份。当所述光固化丙烯酸酯类单体的含量小于所述范围时,即使向由此制备的光固化粘合剂紫外线照射,粘合力减少程度可能微小,而大于所述范围时,过于提高粘合力。
47.在本发明的一实施例中,所述光固化粘合剂组合物可以包含光固化聚氨酯丙烯酸酯类低聚物。其中,所述光固化聚氨酯丙烯酸酯类低聚物可以包括具有2至6个官能团的脂肪族(aliphatic)聚氨酯丙烯酸酯。根据本发明的一实施例,可以使用双官能度脂肪族聚氨酯丙烯酸酯(2-functionality aliphatic urethane acrylate)及/或者六官能度脂肪族聚氨酯丙烯酸酯(6-functionality aliphatic urethane acrylate)。另一方面,当增加所述聚氨酯丙烯酸酯类低聚物的官能团数时,由于高反应性,经紫外线照射后,粘合力急剧减少。
48.在本发明的一实施例中,相对于100重量份的所述丙烯酸共聚物,所述光固化聚氨酯丙烯酸酯类低聚物的含量可以是5重量份至100重量份,优选为20重量份至50重量份,根据本发明的一实施例可以约为30重量份。当所述光固化聚氨酯丙烯酸酯类低聚物的含量大于所述范围时,由于由此制备的光固化粘合剂在照射紫外线前粘合力的内聚力降低,发生内聚破坏,导致粘合力增加,且增加粘合剂在被粘物上的残留量。
49.在本发明的一实施例中,所述光固化粘合剂组合物可以包含交联剂(固化剂)。其中,所述交联剂可以是包括选自2,4-甲苯二异氰酸酯、2,6-甲苯二异氰酸酯、1,5-萘二异氰酸酯、二苯基甲烷二异氰酸酯、六亚甲基二异氰酸酯、异佛尔酮二异氰酸酯、对苯二甲基二异氰酸酯、间苯二甲基二异氰酸酯、二环己基甲烷二异氰酸酯、脂肪族多异氰酸酯及它们的组合中的交联剂。根据本发明的一实施例,所述交联剂可以使用2,4-甲苯二异氰酸酯。所述交联剂的作用在于,在光固化粘合剂组合物聚合时,能够使丙烯酸共聚物、光固化丙烯酸酯类单体及光固化聚氨酯丙烯酸酯类低聚物进行交联,并调节由此聚合的光固化粘合剂的物性。
50.在本发明的一实施例中,相对于100重量份的所述丙烯酸共聚物,所述交联剂的含量可以是0.1重量份至10重量份,优选为0.1重量份至5重量份,根据本发明的一实施例可以约为1重量份。当所述交联剂的含量小于所述范围时,有可能因不能充分地进行固化而残留粘合剂,即,发生内聚破坏,而大于所述范围时,在交联密度提高的同时,粘合力可能减少。
51.在本发明的一实施例中,所述光固化粘合剂组合物可以包含光引发剂。其中,所述光引发剂可以包括选自二苯甲酮、苄基二甲基缩酮、安息香、安息香甲醚、安息香异丙醚、安息香异丁醚、安息香苯甲酸甲酯、安息香苯甲酸、苯乙酮、2,2-二甲氧基-2-苯基苯乙酮、2,4-二乙基噻唑酮、α-羟基环己基苯基酮、苄基二苯硫醚及它们的组合中的物质,优选地,可以包含α-羟基环己基苯基酮。
52.在本发明的一实施例中,相对于100重量份的所述丙烯酸共聚物,所述光引发剂的含量可以是0.1重量份至10重量份,优选为0.5重量份至5重量份,根据本发明的一实施例约为3重量份。当所述光引发剂的含量小于所述范围时,不能很好地进行光引发,从而可能导致无法顺利地进行交联,而大于所述范围时,交联密度增加,导致涂膜变脆(brittle),由此降低对被粘物的粘合力。
53.在本发明的一实施例中,所述光固化粘合剂可以由上述的光固化粘合剂组合物聚合而制备。此时,参照图1,所述聚合可以是通过将所述光固化粘合剂组合物涂布在基材层3而进行,被聚合的光固化粘合剂可以形成第一粘合层1和第二粘合层2。其中,所述聚合可以是在100℃至200℃的温度下干燥1分钟至5分钟而进行,优选为在约120℃的温度下干燥约2分钟。
54.在本发明的一实施例中,所述基材3可以使用可使紫外线(uv)通过的透明或者半透明薄膜,例如,可以使用由选自聚对苯二甲酸乙二酯(polyethylene terephthalate,pet)、聚乙烯(polyethylene,pe)、聚丙烯(polypropylene,pp)、定向聚丙烯(oriented polypropylene,opp)及它们的组合中的物质形成的薄膜。另一方面,所述薄膜的厚度可以是4.5μm至150μm,优选为25μm至75μm。
55.在本发明的一实施例中,所述光固化粘合剂的粘合力可以是3,000gf/25mm以上,优选为3,000gf/25mm至4,000gf/25mm。另一方面,将所述光固化粘合剂涂布在基材后,以100mj/cm2至3,000mj/cm2的光量照射具有200nm至500nm的波长(优选为300nm至400nm的波长)的紫外线时,通过下述数学式1计算出的粘合力减少率为90%以上,优选为98%以上。
56.数学式1:
[0057][0058]
即,所述光固化粘合剂在照射紫外线(uv)后的粘合力可以是50gf/25mm。因此,所述光固化粘合剂可以在照射紫外线前具有高粘合力,从而能够牢牢地保持被粘物的粘合,但是在照射紫外线后,粘合力急剧减少,非常容易从被粘物剥离。
[0059]
另一方面,如图1所示,所述光固化粘合剂可以以基材层3为中心并在其两侧面分别形成第一粘合层1及第二粘合层2,所述第一粘合层1及第二粘合层2的另一侧可以分别形成有第一剥离性薄膜层4和第二剥离性薄膜层5。所述第一剥离性薄膜层4和第二剥离性薄膜层5可以是为了从外部污染物质保护所述光固化粘合剂而形成,具体地,当光固化粘合剂组合物进行光聚合反应时,由于空气中的氧气而使反应受阻,因此为了以将其覆盖的方式防止与氧气接触而使用。之后,可以在实际使用时去除所述第一剥离性薄膜层4和第二剥离性薄膜层5后,粘合在被粘物上。
[0060]
在本发明的一实施例中,所述可再剥离粘合剂可以包括热发泡剂。其中,所述热发泡剂可以是包括选自被微囊化的热膨胀微球、无机发泡剂、有机发泡剂及它们的组合中的物质,优选地,可以使用被微囊化的热膨胀微球。其中,所述被微囊化的热膨胀微球可以使用松本油脂制药株式会社的产品“松本微球”f-30、f-36、f-48、f-80、f-65、fn-100s或者fn-180。另外,考虑到分散性和发泡力,所述热发泡剂的平均粒径可以是1μm至50μm,优选为3μm至30μm。
[0061]
在本发明的一实施例中,相对于100重量份的所述光固化粘合剂,所述热发泡剂的含量可以是0.5重量份至50重量份,优选为10重量份至40重量份,根据本发明的一实施例约为20重量份。当所述热发泡剂的含量小于所述范围时,基于热的膨胀变形可能不充分,导致无法顺利地进行剥离,而大于所述范围时,因膨胀变形充分而易于剥离,但发泡后有可能与基材之间的粘合力降低,导致界面破坏,同时残留有残留物。另一方面,随着所述热发泡剂的含量增加,初期粘合力(tack)降低,因而常温粘合力有可能降低。
[0062]
在本发明的一实施例中,考虑到发泡剂的种类及发泡条件,所述热发泡剂的发泡可以以在100℃至270℃的温度下加热1分钟至5分钟的方式进行。另外,如图1所示,将包含光固化粘合剂及热发泡剂的组合物涂布在基材层3时,涂布厚度可以是10μm至100μm,优选为40μm至70μm。当涂布厚度小于所述范围时,由于热发泡剂粒子而涂布表面可能不均匀,导致对被粘物的湿润性和粘合力降低,而大于所述范围时,从被粘合剂剥离时可能发生问题。
[0063]
本发明一实施例中,所述可再剥离粘合剂的粘合力可以是2,000gf/25mm以上,在紫外线照射及热发泡后,能够完全消除粘合力。因此,可通过紫外线照射及热发泡剥离,而不会在被粘物上残留有残留物。
[0064]
在本发明的一实施例中,上述的同时包含光固化粘合剂及热发泡剂的可再剥离粘合剂,一般在提高粘合剂的固化率时,增加交联密度,从而降低热发泡剂的热膨胀力,因而剥离力升高,难以进行剥离/拆卸。相反,当固化率降低时,交联密度减少,从而增加热膨胀力,由此容易进行剥离/拆卸,但在被粘物上可能残留有粘合残留物。对此,调节所述粘合剂的固化率尤为重要,这个可通过紫外线光量来调节,可测量随光量而变的作为凝胶分率的固化率。其中,关于凝胶分率,根据astm d2765,1)首先,制作一个120目(mesh)的袋子,测量重量,将试样切割成约0.3g后,放入所述袋子内,密封后,重新测量重量;2)使用甲苯(toluene)作为溶剂,萃取12小时;3)将萃取的样品在烤箱中,以150℃的温度干燥15分钟后,测量重量,如下述数学式2所示,计算出凝胶分率。
[0065]
数学式2:
[0066]
凝胶分率(%)=(w3-w1)/(w2-w1)
×
100
[0067]
在所述数学式2中,w1可以是指袋子重量,w2可以是指萃取前装有试样的袋子重量,w3可以是指萃取后袋子的重量。
[0068]
本发明的所述可再剥离粘合剂根据所述数学式2计算的凝胶分率可以是80%至92%,优选约为85%左右。当所述凝胶分率小于80%时,热发泡剂的热膨胀系数为3倍以上,发泡力好,但被粘物上可能残留有粘合残留物,而大于92%时,热发泡剂几乎不发生热膨胀,且发泡时间长。另一方面,当所述凝胶分率约为85%左右时,热发泡剂的热膨胀系数可以恒定为1.5倍至2倍,易于从被粘物剥离,且还不残留粘合残留物。另外,当被照射的紫外线光量小于200mj/cm2时,所述凝胶分率可能小于80%,而大于2,000mj/cm2时,凝胶分率显示为93%以上。
[0069]
在本发明的一实施例中,当从被粘物剥离所述可再剥离粘合剂时,首先,通过紫外线照射而使其固化,从而消除粘合力,其次,通过加热而使热发泡剂膨胀,从而减少与被粘物的表面粘合面积,易于进行脱离(分离)。
[0070]
本发明的第二实施方式提供一种可再剥离胶带,其通过将所述本发明的第一实施方式的可再剥离粘合剂涂布于基材而制备。
[0071]
省略与本发明的第一实施方式重复部分的详细说明,但是即使在第二实施方式中省略其说明,但是关于本发明的第一实施方式的说明内容同样可以适用于第二实施方式。
[0072]
以下,参照图1,对本发明的第二实施方式的所述可再剥离胶带进行详细说明。其中,所述图1是示出本发明的一实施方式的可再剥离胶带的使用状态的概略图。
[0073]
在本发明的一实施例中,所述可再剥离胶带可通过将包括所述光固化粘合剂及热发泡剂的组合物涂布于基材层3而制备。其中,所述涂布厚度可以是10μm至100μm,优选为40
μm至70μm。当涂布厚度小于所述范围时,由于热发泡剂粒子而涂布表面可能不均匀,导致对被粘物的湿润性和粘合力降低,而大于所述范围时,从被粘物再剥离时可能发生问题。另外,所述基材3可以使用可使紫外线通过的透明或者半透明薄膜,例如,可以使用由选自聚对苯二甲酸乙二酯(polyethylene terephthalate,pet)、聚乙烯(polyethylene,pe)、聚丙烯(polypropylene,pp)、定向聚丙烯(oriented polypropylene,opp)及它们的组合中的物质形成的薄膜。另一方面,所述薄膜的厚度可以是4.5μm至150μm,优选为25μm至75μm。
[0074]
另一方面,如图1所示,所述可再剥离胶带可以以基材层3为中心,并在其两侧面分别形成第一粘合层1及第二粘合层2,所述第一粘合层1及第二粘合层2的另一侧可以分别形成有第一剥离性薄膜层4和第二剥离性薄膜层5。所述第一剥离性薄膜层4和第二剥离性薄膜层5可以是为了从外部污染物质保护所述光固化粘合剂而形成,具体地,当光固化粘合剂组合物进行光聚合反应时,由于空气中的氧气而使反应受阻,因此为了以将其覆盖的方式防止与氧气接触而使用。之后,可以在实际使用时去除所述第一剥离性薄膜层4和第二剥离性薄膜层5后,粘合在被粘物上。
[0075]
以下,对本发明的实施例进行详细说明,以便本领域技术人员易于实施。但是本发明能够以各种不同形态实施,而不限于此处说明的实施例。
[0076]
制备例1:丙烯酸共聚物的制备
[0077]
制备例1-1
[0078]
首先,为了制备丙烯酸共聚物,相对于100重量份的由丙烯酸单体即丙烯酸-2-乙基己酯(2-ethylhexyl acrylate,2-eha)、乙酸乙烯酯(vinyl acetate,va)、甲基丙烯酸甲酯(methyl methacrylate,mma)及丙烯酸(acrylic acid,aa)构成的混合物,混合150重量份的作为溶剂的乙酸乙酯(ethyl acetate,ea)及0.3重量份的作为引发剂的2,2偶氮二异丁腈(2,2-azobisisobutyronitrile,aibn)并充分进行搅拌。其中,所述丙烯酸单体的重量比依次为68:20:10:2。将搅拌后的混合物中的1/3加入到四口烧瓶中,吹扫氮气,同时在80℃下反应30分钟后,将剩余2/3在2小时30分钟内滴加的同时进行反应。完成滴加后,保持温度在80℃的同时,进一步反应4小时,从而获得固含量为40%的丙烯酸共聚物。
[0079]
制备例1-2
[0080]
除了将所述制备例1-1中的丙烯酸单体的重量比依次设置为66:20:10:4以外,利用相同的方法来制备丙烯酸共聚物。
[0081]
制备例1-3
[0082]
除了将所述制备例1-1中的丙烯酸单体的重量比依次设置为64:20:10:6以外,利用相同的方法来制备丙烯酸共聚物。
[0083]
将在所述制备例1-1至1-3中分别使用的组成及含量比示于下述表1。
[0084]
表1:制备例1-1至1-3
[0085]
类别制备例1-1制备例1-2制备例1-32-eha686664va202020mma101010aa246ea150150150
aibn0.30.30.3
[0086]
制备例2:光固化粘合剂的制备
[0087]
制备例2-1
[0088]
为了制备光固化粘合剂,首先,将在所述制备例1-1中制备的丙烯酸共聚物、作为单体的1,6-己二醇二丙烯酸酯(1,6-hexanediol diacrylate,miramer m200,美源特殊化工株式会社)、作为低聚物的双官能度脂肪族聚氨酯丙烯酸酯(2-functionality aliphatic urethane acrylate,miramer pu256,美源特殊化工株式会社)、作为交联剂的2,4-甲苯二异氰酸酯(tdi)及作为光引发剂的igacure 184以重量比100:10:30:1:3混合而制备光固化粘合剂组合物。
[0089]
制备例2-2
[0090]
除了在所述制备例2-1中使用六官能度脂肪族聚氨酯丙烯酸酯(6-functionality aliphatic urethane acrylate,miramer pu610,美源特殊化工株式会社)作为低聚物以外,利用相同的方法来制备光固化粘合剂。
[0091]
制备例2-3
[0092]
除了在所述制备例2-1中使用在所述制备例1-2中制备的丙烯酸共聚物以外,利用相同的方法来制备光固化粘合剂。
[0093]
制备例2-4
[0094]
除了在所述制备例2-3中使用六官能度脂肪族聚氨酯丙烯酸酯(6-functionality aliphatic urethane acrylate,miramer pu610,美源特殊化工株式会社)作为低聚物以外,利用相同的方法来制备光固化粘合剂。
[0095]
制备例2-5
[0096]
除了在所述制备例2-1中使用在所述制备例1-3中制备的丙烯酸共聚物以外,利用相同的方法来制备光固化粘合剂。
[0097]
制备例2-6
[0098]
除了在所述制备例2-5中使用六官能脂肪族聚氨酯丙烯酸酯(6-functionality aliphatic urethane acrylate,miramer pu610,美源特殊化工株式会社)作为低聚物以外,利用相同的方法来制备光固化粘合剂。
[0099]
比较制备例1
[0100]
除了在所述制备例2-1中将作为低聚物的六官能度脂肪族聚氨酯丙烯酸酯(6-functionality aliphatic urethane acrylate,miramer pu610,美源特殊化工株式会社)以60重量比混合以外,利用相同的方法来制备光固化粘合剂。
[0101]
比较制备例2
[0102]
除了在所述比较制备例1中使用所述制备例1-2中制备的丙烯酸共聚物以外,利用相同的方法来制备光固化粘合剂。
[0103]
比较制备例3
[0104]
除了在所述比较制备例1中使用所述制备例1-3中制备的丙烯酸共聚物以外,利用相同的方法来制备光固化粘合剂。
[0105]
将在所述制备例2-1至2-6中分别使用的组成及含量比示于下述表2中,并且将在比较制备例1至3中分别使用的组成及含量比显示于下述表3中。
[0106]
表2:制备例2-1至2-6
[0107][0108]
表3:比较制备例1至3
[0109][0110]
实施例:可再剥离胶带的制备
[0111]
实施例1
[0112]
为了制备可再剥离胶带,以100:20的重量比混合所述制备例2-4中制备的光固化粘合剂组合物和作为热发泡剂的f-36d,将其涂布于厚度为50μm的pet薄膜,并在100℃下干燥2分钟,从而制备在各个端面上以50μm的粘合厚度涂布的150μm的两面胶带。
[0113]
另一方面,在随后的实验中照射光量为800mj/cm2(波长365nm,80w)的紫外线(uv)。
[0114]
实施例2
[0115]
除了在所述实施例1中使用f-65作为热发泡剂以外,利用相同的方法来制备两面胶带。
[0116]
实施例3
[0117]
除了在所述实施例1中使用fn-180作为热发泡剂以外,利用相同的方法来制备两面胶带。
[0118]
比较例1
[0119]
除了在所述实施例1中以40重量比混合热发泡剂以外,利用相同的方法来制备两面胶带。
[0120]
比较例2
[0121]
除了在所述实施例2中以40重量比混合热发泡剂以外,利用相同的方法来制备两面胶带。
[0122]
比较例3
[0123]
除了在所述实施例3中以40重量比混合热发泡剂以外,利用相同的方法来制备两面胶带。
[0124]
比较例4
[0125]
利用与所述实施例2相同的方法来制备两面胶带,随后在实验中照射光量为200mj/cm2(波长365nm,80w)的紫外线。
[0126]
比较例5
[0127]
利用与所述实施例2相同的方法来制备两面胶带,随后在实验中照射光量为2,000mj/cm2(波长365nm,80w)的紫外线。
[0128]
将在所述实施例1至3中分别使用的组成、含量比及紫外线光量强度示于下述表4,并且将在比较例1至5分别使用的组成、含量比及紫外线光量强度示于下述表5。
[0129]
表4:实施例1至3
[0130][0131]
表5:比较例1至5
[0132][0133]
实验例1:使用光固化粘合剂的胶带借助于紫外线(uv)照射的粘合力的测量
[0134]
对于在所述制备例2-1至2-6及比较制备例1至3中分别制备的胶带,实施了借助于紫外线照射的粘合力测量实验。为此,首先,在照射紫外线之前测量粘合力,具体地,根据astm d-1000,将试样在sus304上使用2kg辊往返挤压1次,常温下放置20分钟后,利用instron 3343,以剥离角度180
°
、剥离速度300mm/min,测量粘合力。此时,环境温度为23℃,湿度为65%。
[0135]
另外,为了在照射紫外线后测量粘合力,将试样在sus304上使用2kg辊往返挤压1次后,通过安装有6kw金属卤化物灯的紫外线固化装置,以800mj/cm2(波长365nm,80w)光量照射后,利用instron 3343,以剥离角度180
°
、剥离速度300mm/min,测量粘合力。
[0136]
另一方面,为了测量探针的粘性(probe tack),将试样以25
×
25mm大小切割,将测试表面朝向下方并粘贴于环形圈(annular ring)后,利用cheminstrument pt-1000进行测量。此时,环境温度为23℃,湿度为65%。
[0137]
将其结果示于下述表6中,将照射紫外线前后残留于支撑体或被粘物的粘合剂的转移程度如下所示。
[0138]
(
◎
没有转移和污染;
●
没有转移,但有污染;
○
小于10%;
△
小于50%;
×
大于50%)
[0139]
表6:物性评价结果
[0140][0141]
如上表6所示,能够确认随着丙烯酸(aa)含量,即作为极性基团的羧基的量的增加而常温下的粘合力增加,并且能够确认由于在制备例2-1及2-2中的丙烯酸(aa)含量相对少,粘合力的内聚力降低,从而因内聚破坏而增加粘合力。另外,能够确认随着低聚物的官能团数量的增加,粘合力减少,并且能够确认如果低聚物的官能团数量增加,则紫外线固化
时由于高反应性而减少粘合力,且减少幅度大幅增加。另外,能够确认与低聚物的官能团数量无关,当其含量增加时,紫外线照射前粘合剂内聚力降低,从而因内聚破坏而增加内聚力,并且增加粘合剂向被粘物的转移率。
[0142]
实验例2:可再剥离胶带的借助于紫外线(uv)照射的粘合力的测量
[0143]
对于在所述实施例1至3及比较例1至5中分别制备的剥离胶带,实施粘合力测量实验。为此,首先,在常温下测量粘合力,具体地,根据astm d-1000,将试样在sus304上使用2kg辊往返挤压1次,常温下放置20分钟后,利用instron 3343,以剥离角度180
°
、剥离速度300mm/min,测量粘合力。此时,环境温度为23℃,湿度为65%。
[0144]
另外,为了在紫外线照射和热发泡后测量粘合力,将试样在sus304上使用2kg辊往返挤压1次后,首先,通过安装有6kw金属卤化物灯的紫外线固化装置,以800mj/cm2(波长365nm,80w)、200mj/cm2或者2,000mj/cm2光量照射后,其次,在260℃的高温腔室通过加热一分钟来发泡后,利用instron 3343,以剥离角度180
°
、剥离速度300mm/min,测量粘合力。此时,环境温度为23℃,湿度为65%。
[0145]
将其结果示于下述表7中,在紫外线照射及热发泡前后,残留于支撑体或被粘物的粘合剂的转移程度如下所示。
[0146]
(
◎
没有转移和污染;
●
没有转移,但有污染;
○
小于10%;
△
小于50%;
×
大于50%)
[0147]
另一方面,根据astm d2765,如下测量凝胶分率:1)首先,制作一个120目(mesh)的袋子,测量重量,将试样切割成0.3g后,放入所述袋子内,密封后,重新测量重量;2)使用甲苯(toluene)作为溶剂,萃取12小时;3)将萃取的样品在烤箱中,以150℃的温度干燥15分钟后,测量重量,计算凝胶分率。
[0148]
凝胶分率(%)=(w3-w1)/(w2-w1)
×
100
[0149]
w1=袋子重量
[0150]
w2=萃取前装有试样的袋子重量
[0151]
w3=萃取后袋子的重量
[0152]
表7:物性评价结果
[0153]
[0154]
如上表7所示,可以确认随着热发泡剂的含量增加且粒子尺寸增大,粘合力减少。另外,可以确认在一次紫外线照射后,由于光固化而粘合剂内交联度提高,使得基于热的发泡剂膨胀受到阻碍,因此在比实际发泡温度更高的温度下进行发泡,特别是,可以确认fn-180本身没有发泡。
[0155]
实施例1和比较例1中使用的热发泡剂f-360的发泡温度低,因此在用于制备胶带的pet薄膜上涂布粘合剂,且干燥过程中表面上发生部分发泡,从而降低粘合力。
[0156]
综上所述,可以确认实施例2中制备的可再剥离胶带对被粘物的粘合力也优秀,且紫外线照射和发泡后,在没有粘合剂转移的情况下,易于剥离。
[0157]
以上,参照附图与优选实施例对本发明进行详细说明,但本发明的技术思想的范围不限于这些附图和实施例。因此,在本发明的技术思想的范围内,可以存在各种变形例或者等同范围的实施例。因此,本发明的技术思想的权利范围由权利要求书解释,与其相同或等同的范围内的技术思想应都被解释为属于本发明的权利范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1