一种热致变色材料、调光膜及其制备方法和应用与流程
1.本发明涉及光学材料技术领域,尤其涉及一种热致变色材料、调光膜及其制备方法和应用。
背景技术:
2.随着社会发展,生态问题日趋严峻,保护环境和节约能源的重要性已经得到越来越多人的关注和重视。门窗玻璃能耗占建筑能耗的50%,其中节能玻璃能够阻挡太阳光中的热量,减少为稳定室内冷热环境带来的能耗。为了提高能源利用率,防止建筑门窗内侧温度上升,减少室内空调用电,需要门窗玻璃能有效阻挡太阳光中带来热量的红外线。通常这种玻璃采用low
‑
e玻璃、阳光控制镀膜玻璃和涂膜或贴膜玻璃等。low
‑
e玻璃是在玻璃表面镀上多层金属和其他化合物组成的膜系产品,所镀金属膜层通常为ag层,其膜层具有对可见光高透过性以及对近红外线高反射的特性,因此具有优异的隔热效果。但离线low
‑
e玻璃需要做成中空或真空玻璃,通常需要对膜层四周进行铲膜处理。阳光控制镀膜玻璃采用多层金属镀膜,很容易造成光污染。
3.近年来出现了以涂布为主的纳米陶瓷膜,相关技术用纳米金属氧化物材料ito和ato涂布于白玻璃表面形成隔热膜,这些纳米金属氧化物材料具有近红外反射及吸收的作用。还有相关技术将掺杂钨铈锑的二氧化锡与eva树脂混合制成胶膜,利用胶膜阻隔紫外线和红外线。另有相关技术先在pet层上溅射一层溅射层,然后涂布一层三氧化钨隔热层,接着复合一层pet层,再涂布一层由聚丙烯酸酯树脂和紫外线吸收剂组成的安装层,最后复合经表面处理的聚酯薄膜层。此隔热膜层具有优异的可见光透过率及红外和紫外阻隔率。也有相关技术将红外阻隔材料分散液与pvb混合,使得胶片在可见光透射率高的同时,又能很好的吸收和反射红外线。以上这些技术均因膜层对红外线有一定的反射性或吸收性而具有隔热效应,其中大多数以吸收近红外线为主,这样容易造成吸收在玻璃表面的热量达到饱和而引起二次热传递,进而起不了隔热作用。
技术实现要素:
4.本发明旨在至少解决现有技术中存在的技术问题之一。为此,本发明提出一种热致变色材料,能够在受热时变色,降低透光率,对太阳光进行有效遮挡。
5.同时,本发明还提供一种调光膜,能使可见光透过同时又能有效阻止红外线通过玻璃进入室内,特别在炎热的夏天能有效阻止热量进入,减少为稳定室内冷热环境带来的能耗,起到节能的目的。
6.同时,本发明还提供所述热致变色材料和调光膜的制备方法和应用。
7.具体地,本发明采取如下的技术方案:
8.本发明的第一方面是提供一种热致变色材料,所述热致变色材料含有铜螯合物和含羟基聚合物,所述含羟基聚合物包覆所述铜螯合物。
9.根据本发明第一方面提供的热致变色材料,至少具有如下有益效果:
10.发明人发现,以铜螯合物作为内容物,采用含羟基聚合物对其进行包覆,所得材料为透明材料。在温度升高,例如由太阳光产生的热量引起温度升高时,铜螯合物中的铜离子与聚合物中的羟基作用产生深蓝色的cu(oh)2凝胶簇,使得膜层状态和颜色发生改变,从而引起透光率的降低,对太阳光进行有效遮挡,达到减弱光线的目的。当太阳光强度减弱,热致变色材料又恢复变色前的状态。
11.在本发明的一些实施方式中,所述含羟基聚合物和铜螯合物的质量比为5~30:1,例如可选5~20:1、5~15:1、5~10:1、10~30:1、15~30:1、10~20:1、15~20:1等。
12.在本发明的一些实施方式中,所述含羟基聚合物的分子量为200~2000。
13.在本发明的一些实施方式中,所述含羟基聚合物包括聚乙二醇、聚对苯二甲酸乙二醇酯、聚对苯二甲酸丁二醇酯、聚碳酸酯、聚乳酸、聚丁二酸丁二醇酯、聚甘油、淀粉中的任意一种或多种的组合,优选聚乙二醇。
14.在本发明的一些实施方式中,所述铜螯合物包括柠檬酸螯合铜、丙氨酸螯合铜、谷氨酸螯合铜、edta
‑
cu中的任意一种或多种的组合,优选柠檬酸螯合铜。
15.本发明的第二方面是提供所述热致变色材料的制备方法,包括如下步骤:将铜螯合物分散在溶剂中,得到铜螯合物分散液,然后与含羟基聚合物混合,得到热致变色材料。所述溶剂包括1,6
‑
己二醇二丙烯酸酯(hdda)、三羟甲基丙烷三甲基丙烯酸酯(tmptma)以及三烯丙基异三聚氰酸酯(taic)、双环戊二烯乙氧基甲基丙烯酸酯、丙烯酰吗啉中的任意一种或多种的组合。
16.在将铜螯合物分散在溶剂过程中,可视情况加入分散剂。所述分散剂可采用通用的分散剂,例如聚乙烯吡咯烷酮、聚丙烯酸钠、多偏磷酸钠等,优选聚乙烯吡咯烷酮。所述聚乙烯吡咯烷酮的分子量为40000~60000。所述分散剂的添加量为铜螯合物质量的10%~50%。
17.在本发明的一些实施方式中,所述铜螯合物分散液中,铜螯合物的质量浓度为1%~10%。
18.本发明的第三方面是提供一种热致变色组合物,所述热致变色组合物含有上述热致变色材料。
19.在本发明的一些实施方式中,所述热致变色组合物的原料包括:
20.弹性体材料
21.所述热致变色材料的分散液
22.交联剂
23.偶联剂
24.助剂。
25.将热致变色材料与弹性体材料以及各种助剂一起制成组合物,可用于制作具有热致变色功能的薄膜。其中热致变色材料以分散液的形式加入组合物中,有助于其中铜离子与羟基的水解和醇解。
26.在本发明的一些实施方式中,所述热致变色组合物包括如下质量份的原料:
27.弹性体材料100份
28.热致变色材料的分散液0.1~10份
29.交联剂0.1~1份
30.偶联剂0.1~1份
31.助剂0.1~1.5份。
32.在本发明的一些实施方式中,所述助剂包括紫外线吸收剂、光稳定剂、抗氧剂中的一种或几种的组合,优选包括紫外线吸收剂、光稳定剂、抗氧剂的组合。
33.在本发明的一些实施方式中,所述热致变色组合物包括如下质量份的原料:
34.弹性体材料100份
35.热致变色材料的分散液0.1~10份
36.交联剂0.1~1份
37.偶联剂0.1~1份
38.紫外线吸收剂0.1~0.5份
39.光稳定剂0.01~0.3份
40.抗氧剂0.01~0.3份。
41.在本发明的一些实施方式中,所述热致变色组合物中,弹性体材料的熔融指数为20~50g/10min(190℃/2.16kg)。
42.在本发明的一些实施方式中,所述弹性体材料包括eva、sbs、poe、sis、氢化sis、tpu、pp中的任意一种或几种的组合,优选包括eva。所述eva可选择日本三井eva
‑
150、韩国韩华282pv、新加坡tpc ka
‑
40、新加坡tpc ka
‑
10、韩国lg28025中的任意一种或多种的组合。
43.在本发明的一些实施方式中,所述热致变色材料的分散液中,热致变色材料的质量浓度为15%~40%。
44.本发明的第四方面是提供一种所述热致变色组合物的制备方法,包括如下步骤:将弹性体材料、热致变色材料的分散液、交联剂、偶联剂和助剂混合,得到热致变色组合物。
45.在本发明的一些实施方式中,在将所述弹性体材料与其他原料混合前,还包括对所述弹性体材料进行冷冻、破碎的步骤。所述冷冻的温度为
‑
50~
‑
30℃。破碎后,所述弹性体材料的粒径为100~500目,优选100~350目。
46.更具体地,所述热致变色组合物的制备方法包括如下步骤:
47.将铜螯合物和溶剂混合(视情况加入分散剂),超声(频率800~3000hz)分散10~30min,得到铜螯合物分散液;
48.将含羟基聚合物与所述铜螯合物分散液混合,搅拌得到热致变色材料的分散液;
49.将弹性体材料在
‑
50~
‑
30℃下冷冻、粉碎至100~350目,然后与热致变色材料的分散液、交联剂、偶联剂和助剂混合,搅拌、干燥得到热致变色组合物。所述搅拌的时间为5~30min,干燥的温度为40~60℃。
50.本发明的第五方面是提供一种隔热材料,含有上述热致变色组合物。
51.本发明的第六方面是提供上述热致变色组合物在制作夹层玻璃中的应用。
52.本发明的第七方面是提供一种调光膜,所述调光膜包括依次层叠的热致变色层、上转换层和反射层,所述热致变色层含有上述热致变色组合物,所述上转换层含有ce
x
y
y
cs
z
w
m
o3,其中x:y:z:m=0.0005~0.1:0.0005~0.1:0.1~0.5:1,优选x:y:z:m=0.001~0.1:0.001~0.05:0.1~0.4:1,更优选x:y:z:m=0.001~0.05:0.001~0.03:0.33:1。
53.根据本发明的调光膜,至少具有如下有益效果:
54.本发明的调光膜由热致变色层、上转换层和反射层组成,其中反射层可以对太阳光进行反射,初步减少太阳光在调光膜中的透过。ce
x
y
y
cs
z
w
m
o3,即ce、y共掺杂cs
z
w
m
o3纳米陶瓷材料,能够吸收近红外线,同时具有上转换功能,将波长更长的红外线,尤其是近红外线转换为波长较短的可见光。因此,未被反射的剩余光线中的部分红外线被上转换层吸收,或者转换为可见光,从而减少红外线在调光膜中的透过率,进一步隔热,且又能增强可见光的透过。最后,未被反射、吸收或转换的光线到达热致变色层,使热致变色层温度升高,然后发生变色,导致热致变色层的透光率降低,从而对可见光和红外线进行遮挡;而当太阳光强度变弱,热致变色层又恢复到透明清爽状态。因此,通过热致变色层、上转换层和反射层的共同作用,充分对光谱进行选择性吸收、透射与反射的同时,将红外线转换为可见光,最后又通过热致变色层阻挡太阳光,起到智能“窗帘”的作用,真正达到隔热节能的目的。
55.本发明还提供另一种调光膜,所述调光膜包括依次层叠的热致变色层和上转换层,所述热致变色层含有上述热致变色组合物,所述上转换层含有ce
x
y
y
cs
z
w
m
o3,其中x:y:z:m=0.0005~0.1:0.0005~0.1:0.1~0.5:1,优选x:y:z:m=0.001~0.1:0.001~0.05:0.1~0.4:1,更优选x:y:z:m=0.001~0.05:0.001~0.03:0.33:1。
56.或者,所述调光膜包括依次层叠的热致变色层和反射层,所述热致变色层含上述热致变色组合物。
57.在本发明的一些实施方式中,所述ce
x
y
y
cs
z
w
m
o3的d50为20~80nm,优选30~60nm,更优选40~50nm。
58.在本发明的一些实施方式中,所述ce
x
y
y
cs
z
w
m
o3的制备方法为:将钨盐和铯盐混合反应后与铈盐、钇盐混合,反应得到前驱体;将所述前驱体煅烧得到ce、y共掺杂cs
z
w
m
o3纳米陶瓷材料,即ce
x
y
y
cs
z
w
m
o3。
59.在本发明的一些实施方式中,所述钨盐、铯盐、铈盐和钇盐以溶液的形式混合,钨盐和铯盐的反应温度,以及与铈盐、钇盐混合后的反应温度不高于用于溶解钨盐、铯盐、铈盐和钇盐的溶剂的沸点。所述溶剂包括水、小分子醇溶剂(如甲醇、乙醇、异丙醇、正丁醇)等。例如,当采用水为溶剂时,钨盐和铯盐的反应温度,以及与铈盐、钇盐混合后的反应温度独立地为60~100℃;采用乙醇为溶剂时,钨盐和铯盐的反应温度,以及与铈盐、钇盐混合后的反应温度独立地为60~78℃,优选70~78℃。为避免钨盐、铯盐、铈盐和钇盐水解,所述溶剂优选小分子醇溶剂。
60.在本发明的一些实施方式中,所述煅烧的温度为400~800℃,优选400~600℃;煅烧的时间为2~5小时,优选3~5小时。所述煅烧过程中通入氮气和氢气的混合气体,所述氮气和氢气的体积流量比为2~5:1。
61.在本发明的一些实施方式中,所述ce
x
y
y
cs
z
w
m
o3的制备方法具体为:将铯盐溶液加入钨盐溶液中,反应得到混合液;然后将铈盐溶液和钇盐溶液与所述混合液混合,反应得到前驱体;将所述前驱体煅烧得到ce
x
y
y
cs
z
w
m
o3。
62.更具体地,所述ce
x
y
y
cs
z
w
m
o3的制备方法为:对钨盐溶液进行回流搅拌1~10小时,然后在保持回流搅拌下将铯盐溶液以1~5滴/秒的速度缓慢滴加到钨盐溶液中,待铯盐溶液滴加完毕后,同时滴加铈盐溶液和钇盐溶液并保持速度为1~3滴/秒,待铈盐溶液和钇盐溶液滴加完毕后继续加热回流搅拌1~10小时;然后冷却至室温并静置陈化10~30小时得到溶胶;将所述溶胶减压蒸馏直到得到透明湿凝胶,干燥并粉碎后得到前驱体粉末;再将前
驱体粉末在400~800℃(优选400~600℃)煅烧2~5小时(优选3~5小时),并通入氮气和氢气混合气体,氮气流量为0.2~1l/min,氢气流量为0.1~0.6l/min,煅烧结束后得到ce
x
y
y cs
z
w
m
o3。其中,回流的温度即前述的钨盐和铯盐的反应温度,以及与铈盐、钇盐混合后的反应温度,不高于铯盐溶液、钨盐溶液、铈盐溶液和钇盐溶液的溶剂的沸点。
63.在本发明的一些实施方式中,所述钨盐包括六氯化钨、三氯化钨、五氯化钨、钨酸钠、钨酸钾、钨酸铵以及这些钨盐的水合物中的任意一种或多种,优选包括六氯化钨或其水合物。所述铯盐包括氯化铯、硝酸铯、氟化铯、硫酸铯、碳酸铯以及这些铯盐的水合物中的任意一种或多种,优选包括氯化铯或其水合物。所述铈盐包括硝酸铈、硫酸铈、氯化铈以及这些铈盐的水合物中的任意一种或多种,优选包括硝酸铈或其水合物。所述钇盐包括硝酸钇、硫酸钇、氯化钇以及这些钇盐的水合物中的任意一种或多种,优选包括硝酸钇或其水合物。
64.在本发明的一些实施方式中,所述铈盐、钇盐、铯盐和钨盐中的铈、钇、铯、钨四种元素的摩尔比为0.0005~0.1:0.0005~0.1:0.1~0.5:1,优选0.001~0.1:0.001~0.05:0.1~0.4:1,更优选0.001~0.05:0.001~0.03:0.33:1。
65.在本发明的一些实施方式中,所述上转换层由上转换组合物形成,所述上转换组合物的原料包括:
66.弹性体材料
67.ce
x
y
y
cs
z
w
m
o368.交联剂
69.偶联剂
70.助剂。
71.在本发明的一些实施方式中,所述上转换组合物包括如下质量份的原料:
72.弹性体材料100份
73.ce
x
y
y
cs
z
w
m
o
3 0.05~0.75份
74.交联剂0.1~1份
75.偶联剂0.1~1份
76.助剂0.1~1.5份。
77.在本发的一些实施方式中,所述助剂包括紫外线吸收剂、光稳定剂、抗氧剂中的一种或几种的组合,优选包括紫外线吸收剂、光稳定剂、抗氧剂的组合。
78.在本发明的一些实施方式中,所述上转换组合物包括如下质量份的原料:
79.弹性体材料100份
80.ce
x
y
y
cs
z
w
m
o
3 0.05~0.75份
81.交联剂0.1~1份
82.偶联剂0.1~1份
83.紫外线吸收剂0.1~0.5份
84.光稳定剂0.01~0.3份
85.抗氧剂0.01~0.3份。
86.在本发明的一些实施方式中,所述上转换组合物的原料中,弹性体材料的熔融指数较热致变色组合物中的更低,具体为1~10g/10min(190℃/2.16kg)。上转换层所使用的弹性体材料的熔融指数相较热致变色层中的更低,硬度会更高,从而可避免上转换层与热
致变色层的原料混在一起,避免难以分层。
87.在本发明的一些实施方式中,所述上转换组合物的原料中,所述弹性体材料包括eva、sbs、poe、sis、氢化sis、tpu、pp中的任意一种或几种的组合,优选为eva。所述eva可选择日本三井eva
‑
260、韩国lg28005、泰国mv1055、新加坡tpc ka
‑
31中的任意一种或多种的组合。
88.在本发明的一些实施方式中,所述ce
x
y
y
cs
z
w
m
o3以分散液的形式加入所述上转换组合物中,所述ce
x
y
y
cs
z
w
m
o3分散液中ce
x
y
y
cs
z
w
m
o3的固含量为10%~30%,ce
x
y
y
cs
z
w
m
o3分散液在上转换组合物中的质量份为0.5~2.5份。所述ce
x
y
y
cs
z
w
m
o3分散液含有ce
x
y
y
cs
z
w
m
o3、分散剂、溶剂。其中,所述分散剂的质量为ce
x
y
y
cs
z
w
m
o3的33.3%~62.5%;所述分散剂可选用通用的分散剂,例如毕克byk
‑
2200、尚高sago9311、埃夫卡efka4310、尚高sago
‑
9105等。所述溶剂包括环己烷、甲基异丁基酮、甲乙酮、丙二醇甲醚醋酸酯、乙酸乙酯中的任意一种或多种的组合。
89.更具体地,所述ce
x
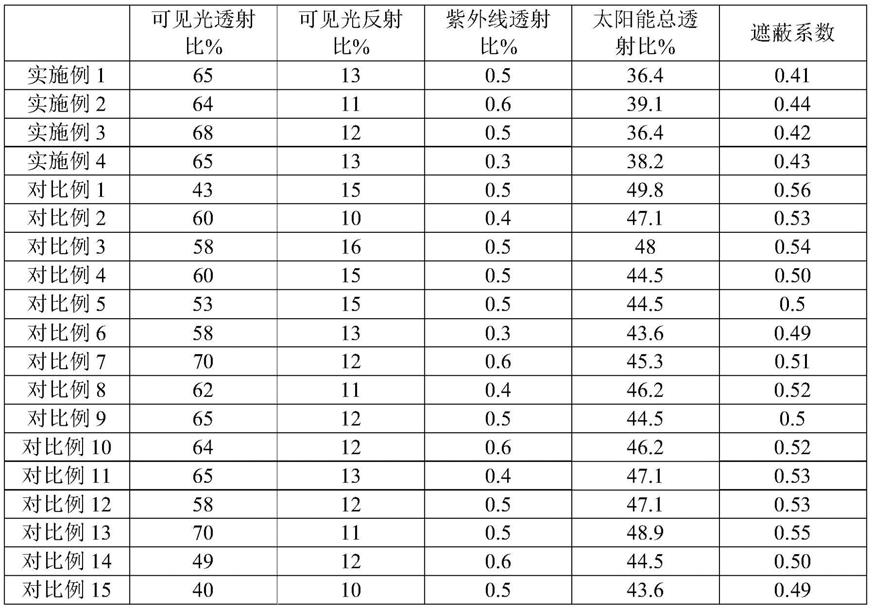
y
y
cs
z
w
m
o3分散液可按如下方法制得:将分散剂、溶剂混合,加入ce
x
y
y
cs
z
w
m
o3,搅拌均匀后超声波分散5~30min,得到悬浊液;采用0.1~0.3mm的研磨介质对所述悬浊液进行研磨,得到ce
x
y
y
cs
z
w
m
o3分散液。研磨过程中控制温度25~40℃,转速为1800~2800r/min,研磨时间为2~5小时。
90.在本发明的一些实施方式中,所述上转换组合物的制备方法为,将弹性体材料、ce
x
y
y cs
z
w
m
o3、交联剂、偶联剂和助剂混合,得到上转换组合物。
91.在本发明的一些实施方式中,所述调光膜的反射层含有纳米银线复合纳米陶瓷材料,所述纳米银线复合纳米陶瓷材料具有纳米银线包裹纳米陶瓷的结构。
92.相关技术一般采用纳米银颗粒与纳米陶瓷进行复合用于制作反射层,由于纳米银颗粒容易被纳米陶瓷包裹,在受到纳米陶瓷阻隔情况下难以发挥反射红外线的作用。本发明在反射层中采用纳米银线代替纳米银颗粒,纳米银线能够避免被纳米陶瓷包裹覆盖,有效发挥反射作用;而且相较纳米银颗粒,纳米银线的镜面反射作用更强。
93.在本发明的一些实施方式中,所述纳米银线复合纳米陶瓷材料的制备原料包括:纳米银线、纳米陶瓷。所述纳米银线与纳米陶瓷的质量比为1:2~6。
94.在本发明的一些实施方式中,所述纳米银线的线径为10~50nm,优选10~20nm;所述纳米银线的线长为5~50μm,优选15~40μm,更优选10~35μm。
95.在本发明的一些实施方式中,所述纳米陶瓷的d90为10~50nm。
96.在本发明的一些实施方式中,所述纳米陶瓷包括氧化铟锡(ito)、氧化锡锑(ato)、掺氟氧化锡(fto)、掺铝氧化锌(azo)中的任意一种或多种的组合,优选氧化铟锡。
97.所述ito中,sn与in的摩尔比为1:5~15,优选1:8~12,更优选约1:10;所述ito的d90为15~30nm。所述ato中,sb与sn的摩尔比为1:5~15,优选1:8~12,更优选约1:10;所述ato的d90为25~40nm。所述fto中,f与sn的摩尔比为1:10~30,优选1:15~25,更优选约1:20;所述fto的d90为10~40nm。所述azo中,al与zn的摩尔比为1:10~30,优选1:15~25,更优选约1:20;所述azo的d90为25~40nm。
98.在本发明的一些实施方式中,所述纳米银线复合纳米陶瓷材料的制备方法包括如下步骤:将纳米银线与纳米陶瓷混合,反应得到纳米银线复合纳米陶瓷材料。
99.更具体地,所述纳米银线复合纳米陶瓷材料的制备方法为:将纳米银线分散于溶
剂中,搅拌并回流1~3小时,然后向其中加入纳米陶瓷,继续搅拌回流8~10小时,去除溶剂、干燥得到纳米银线复合纳米陶瓷材料。其中,去除溶剂的方法可采用减压蒸馏、加热挥发法等。所述搅拌、回流的温度不高于所述溶剂的沸点。所述溶剂包括水、乙醇、甲醇、异丙醇、正丁醇等。所述搅拌、回流的温度优选为50~78℃,更优选70~78℃。
100.在将纳米银线分散于溶剂过程中,可视情况加入分散剂。所述分散剂可采用通用的分散剂,例如聚乙烯吡咯烷酮、kh
‑
570(γ
‑
甲基丙烯酰氧基丙基三甲氧基硅烷)等,优选聚乙烯吡咯烷酮。所述聚乙烯吡咯烷酮的分子量为40000~60000。所述分散剂的添加量为纳米银线与纳米陶瓷总质量的15%~50%。
101.在本发明的一些实施方式中,所述反射层由反射组合物形成,所述反射组合物的原料包括:
102.弹性体材料
103.纳米银线复合纳米陶瓷材料
104.交联剂
105.偶联剂
106.助剂。
107.在本发明的一些实施方式中,所述反射组合物包括如下质量份的原料:
108.弹性体材料100份
109.纳米银线复合纳米陶瓷材料0.01~0.24份
110.交联剂0.1~1份
111.偶联剂0.1~1份
112.助剂0.1~1.5份。
113.在本发的一些实施方式中,所述助剂包括紫外线吸收剂、光稳定剂、抗氧剂中的一种或几种的组合,优选包括紫外线吸收剂、光稳定剂、抗氧剂的组合。
114.在本发明的一些实施方式中,所述反射组合物包括如下质量份的原料:
115.弹性体材料100份
116.纳米银线复合纳米陶瓷材料0.01~0.24份
117.交联剂0.1~1份
118.偶联剂0.1~1份
119.紫外线吸收剂0.1~0.5份
120.光稳定剂0.01~0.3份
121.抗氧剂0.01~0.3份。
122.在本发明的一些实施方式中,所述反射组合物的原料中,弹性体材料的熔融指数为20~50g/10min(190℃/2.16kg)。
123.在本发明的一些实施方式中,所述反射组合物的原料中,所述弹性体材料包括eva、sbs、poe、sis、氢化sis、tpu、pp中的任意一种或几种的组合,优选为eva。所述eva可选择日本三井eva
‑
150、韩国韩华282pv、新加坡tpc ka
‑
40、新加坡tpc ka
‑
10、韩国lg28025中的任意一种或多种的组合。
124.在本发明的一些实施方式中,所述纳米银线复合纳米陶瓷材料以分散液的形式加入所述反射组合物中,所述纳米银线复合纳米陶瓷材料的分散液中,纳米银线复合纳米陶
瓷材料的固含量为5%~16%,分散液在反射组合物中的质量份为0.2~1.5份。所述纳米银线复合纳米陶瓷材料的分散液含有纳米银线复合纳米陶瓷材料、分散剂和溶剂。其中,所述分散剂的添加量为纳米银线复合纳米陶瓷材料质量的33.3%~62.5%;所述分散剂可选用通用的分散剂,例如毕克byk
‑
2200、尚高sago9311、埃夫卡efka4310、尚高sago
‑
9105等。所述溶剂包括无水乙醇、异丙醇、正丁醇中的任意一种或多种的组合。
125.具体地,所述纳米银线复合纳米陶瓷材料的分散液可按如下方法制得:将分散剂、溶剂混合,加入纳米银线复合纳米陶瓷材料,搅拌均匀后超声波分散5~30min,得到悬浊液;采用0.2~0.5mm的研磨介质对所述悬浊液进行研磨,得到纳米银线复合纳米陶瓷材料的分散液,研磨过程中控制温度25~40℃,转速为1200~2000r/min,研磨时间为1~3小时。
126.需要说明的是,所述反射组合物、上转化组合物以及热致变色组合物中的交联剂、偶联剂、光稳定剂、抗氧剂可以相同也可以不同。所述反射组合物与热致变色组合物中的eva可以采用相同或不同牌号、熔融指数的产品。
127.具体地,所述反射组合物、上转化组合物以及热致变色组合物中,所述交联剂独立地包括2,5
‑
二甲基
‑
2,5
‑
二(2
‑
乙基己酰过氧化)己烷、1,1
‑
二叔丁基过氧化
‑
3,3,5
‑
甲基环己烷、1,1
‑
二特戊基过氧化环己烷、过氧异丁酸叔丁酯中的任意一种或多种的组合,其他通用的交联剂也适用。
128.所述偶联剂独立地包括γ
‑
氨丙基三乙氧基硅烷、γ
‑
甲基丙烯酰氧基丙基三甲氧基硅烷、乙烯基三甲氧基硅烷、氨丙基三甲氧基硅烷中的任意一种或多种的组合。
129.所述紫外线吸收剂独立地包括2
‑
羟基
‑4‑
正辛氧基二苯甲酮、2
‑
(2'
‑
羟基
‑
3',5'
‑
二叔戊基苯基)苯并三唑、2
‑
(4,6
‑
双(2,4
‑
二甲基苯基)
‑
1,3,5
‑
三嗪
‑2‑
基)
‑5‑
辛氧基酚、2
‑
(2'
‑
羟基
‑
5'
‑
特辛基)苯基苯并三氮唑、纳米氧化锌(d90为10~30nm)、纳米氧化铈(d90为15~50nm)中的任意一种或多种的组合,其他通用的紫外线吸收剂也适用。
130.所述光稳定剂独立地包括双(1,2,2,6,6
‑
五甲基
‑4–
哌啶基)癸二酸酯、双(2,2,6,6
‑
四甲基
‑4‑
哌啶基)癸二酸酯、聚丁二酸(4
‑
羟基
‑
2,2,6,6
‑
四甲基
‑1‑
哌啶乙醇)酯中的任意一种或多种的组合,其他通用的光稳定剂也适用。
131.所述抗氧剂独立地包括β
‑
(3,5
‑
二叔丁基
‑4‑
羟基苯基)丙酸十八碳醇酯、季戊四醇四(3
‑
月桂基硫代丙酸酯)、三(2,4
‑
二叔丁基苯基)亚磷酸酯、抗氧剂1098、抗氧化1010、抗氧化1076、抗氧化ca、抗氧剂164中的任意一种或多种的组合;所述抗氧剂优选为β
‑
(3,5
‑
二叔丁基
‑4‑
羟基苯基)丙酸十八碳醇酯与季戊四醇四(3
‑
月桂基硫代丙酸酯)的混合物,二者的质量比为1~2:1。
132.在本发明的一些实施方式中,所述反射组合物的制备方法为:将弹性体材料、纳米银线复合纳米陶瓷材料、交联剂、偶联剂和助剂混合,搅拌3~30min,得到反射组合物。
133.在本发明的一些实施方式中,所述调光膜中,所述热致变色层、上转换层和反射层的厚度分别独立为0.1~1mm,优选独立为0.15~0.4mm。
134.本发明还提供所述调光膜的制备方法,包括如下步骤:将所述反射组合物、上转换组合物以及热致变色组合物进行流延、挤出、冷却、成型,得到调光膜。
135.更具体地,所述调光膜的生产方法为:将反射组合物、上转换组合物以及热致变色组合物分别放在流延挤出机的三个进料斗中,控制三条螺杆机筒各温区的温度50~75℃,弯头及连接采用水循环冷却,温度控制为45~60℃,换网器温度控制为48~60℃,模头各温
区控制为70~85℃,冷冻机胶辊温度控制为10~15℃,冷冻机铁辊温度控制为20~25℃,通过分配器,流延、挤出、冷却、成型,得到调光膜。
136.当所述调光膜仅包括依次层叠的热致变色层和上转换层,或者仅包括依次层叠的热致变色层和反射层时,则在前述的制备方法中相应地省去反射组合物或上转换组合物即可,其他操作相同。
137.本发明还提供一种夹层玻璃,所述夹层玻璃包括上述调光膜。
138.在本发明的一些实施方式中,所述夹层玻璃包括依次层叠的第一玻璃层、调光膜、第二玻璃层。所述第一玻璃层和第二玻璃层可独立地采用普白玻璃、超白玻璃或者其他常用玻璃。
139.本发明还提供所述夹层玻璃在建筑门窗或玻璃幕墙中的应用。
140.相对于现有技术,本发明具有如下有益效果:
141.本发明采用流延挤出机将三种不同功能的组合物通过三层共挤流延挤出成型为具有三层结构的智能调光膜,三层结构包括下层热致变色层,中间层为具有上转换功能的上转换层,最上层为纳米银线复合纳米陶瓷材料制成的反射层。当太阳光照射过来后,反射层中的纳米银线反射大部分红外线,复合的具有隔热作用的纳米陶瓷同时阻隔近红外线。当反射层没有阻隔的红外线经过中间的上转换层后,上转换层在吸收近红外线的同时又能将部分近红外线转换为可见光,从而在保证进一步隔热的同时,又能增强可见光的透过。当夏季天气炎热的情况下,最上面两层不足以阻挡太阳光的热量时,太阳光产生的热量使得热致变色层中的铜螯合物的铜离子在聚合物的羟基的作用下形成深蓝色的cu(oh)2凝胶簇,使得热致变色层的膜层状态和颜色发生改变,阻挡太阳光进入室内,进一步达到隔热节能的目的,而当太阳光强度变弱,热致变色层的膜层又恢复到透明清爽状态。本发明三层共挤智能调光膜,充分结合膜系对光谱选择性吸收、透射与反射的同时,辅助上转换将红外线转换为可见光,最后又通过热致变色阻挡太阳光,起到智能“窗帘”的作用,真正达到隔热节能的目的。
附图说明
142.图1为实施例1的ce、y共掺杂cs
0.33
wo3纳米陶瓷粉体在不同放大倍数下的sem图;
143.图2为实施例1的ce、y共掺杂cs
0.33
wo3纳米陶瓷粉体的xrd图;
144.图3为实施例1的夹层玻璃的uv
‑
vis
‑
nir透射与正反面反射光谱图;
145.图4为实施例1的夹层玻璃在980nm激发光下的发射光谱;
146.图5为实施例1的夹层玻璃在808nm激发光下的发射光谱。
具体实施方式
147.以下结合具体的实施例进一步说明本发明的技术方案。以下实施例中所用的原料,如无特殊说明,均可从常规商业途径得到;所采用的工艺,如无特殊说明,均采用本领域的常规工艺。
148.实施例1
149.一种三层共挤智能调光eva膜,包括依次层叠的下层、中间层和上层,其中,下层为热致变色eva膜,中间层为上转换纳米陶瓷eva膜,上层为纳米银线复合纳米陶瓷eva膜。三
层共挤智能调光eva膜的制备方法包括如下步骤:
150.s1:下层热致变色组合物,即热致变色eva膜功能预备料的制备
151.称取25g柠檬酸螯合铜放在500ml烧杯中,向其中加入10g聚乙烯吡咯烷酮,再滴加215g的1,6
‑
己二醇二丙烯酸酯,搅拌均匀后利用超声波细胞粉碎仪超声分散30min,超声波频率为2000hz,得到柠檬酸螯合铜分散液。再将125g peg(分子量为1250)逐滴滴加到柠檬酸螯合铜分散液中,边滴加边搅拌均匀使柠檬酸螯合铜表面完全包覆peg,得到柠檬酸螯合铜/peg的分散液。
152.称取5kg日本三井eva
‑
150颗粒,在
‑
40℃下冷冻粉碎至350目,然后称取250g柠檬酸螯合铜/peg的分散液、50g 1,1
‑
二叔丁基过氧化
‑
3,3,5
‑
甲基环己烷、50gγ
‑
甲基丙烯酰氧基丙基三甲氧基硅烷、20g 2
‑
羟基
‑4‑
正辛氧基二苯甲酮、10g双(1,2,2,6,6
‑
五甲基
‑4‑
哌啶基)癸二酸酯、10gβ
‑
(3,5
‑
二叔丁基
‑4‑
羟基苯基)丙酸十八碳醇酯和5g季戊四醇四(3
‑
月桂基硫代丙酸酯)混合均匀加入到eva粉末中搅拌均匀并干燥,干燥温度为40℃,搅拌时间为25min,得到下层热致变色eva膜功能预备料。
153.s2:中间层上转换组合物,即上转换纳米陶瓷eva膜功能预备料的制备
154.s21:ce、y共掺杂cs
0.33
wo3纳米陶瓷(ce
0.05
y
0.03
cs
0.33
wo3)粉体制备
155.称取39.65g(0.1mol)六氯化钨于500ml烧杯中,加200ml无水乙醇溶解得到a液;称取5.56g(0.033mol)氯化铯于50ml烧杯中,加30ml无水乙醇溶解得到b液;称取2.17g(0.005mol)六水合硝酸铈于50ml烧杯中,加20ml无水乙醇溶解得到c液;称取1.15g(0.003mol)六水合硝酸钇于50ml烧杯中,加20ml无水乙醇溶解得到d液。
156.然后将a液转入到500ml四口烧瓶中,于76℃回流搅拌3小时,继续回流搅拌并将b液、c液以及d液分别转入到50ml恒压滴液漏斗中。具体地,先将b液以3滴/秒速度缓慢滴加到a液中,待b液滴加完毕后,同时滴加c液和d液并保持速度为2滴/秒,待c液、d液滴加完毕后继续加热回流搅拌3小时,然后冷却至室温并静置陈化24小时得到溶胶。
157.将溶胶减压蒸馏直到得到透明湿凝胶,干燥并粉碎后得到前驱体粉末。再将前驱体粉末在马弗炉中500℃煅烧5h,并通入氮气和氢气混合气体,氮气流量为0.8l/min,氢气流量为0.4l/min,最终得到ce、y共掺杂cs
0.33
wo3纳米陶瓷粉体,研磨称重24.75g,其sem图、xrd图分别如图1和图2所示。ce、y共掺杂cs
0.33
wo3纳米陶瓷粉体含有较规则的的立方体颗粒形貌,立方体颗粒的粒径为49nm(d50)。
158.s22:ce、y共掺杂cs
0.33
wo3纳米陶瓷分散液的制备
159.称取10g毕克byk
‑
2200分散剂于250ml烧杯中,向其中加入70g甲基异丁基酮,搅拌均匀直至分散剂完全溶解于溶剂中,再向混合液中加入20g ce、y共掺杂cs
0.33
wo3纳米陶瓷粉体,搅拌均匀,然后超声波分散10min,最后将悬浊液转入0.3l棒硝式纳米砂磨机,并加入0.2mm镐珠,控制物料温度25~40℃,转速为2800r/min,砂磨时间为5小时得到ce、y共掺杂cs
0.33
wo3纳米陶瓷分散液。
160.s23:中间层上转换纳米陶瓷eva膜功能预备料的制备
161.称取80g的ce、y共掺杂cs
0.33
wo3纳米陶瓷分散液,向其中加入50g 1,1
‑
二叔丁基过氧化
‑
3,3,5
‑
甲基环己烷交联剂、50gγ
‑
甲基丙烯酰氧基丙基三甲氧基硅烷、20g 2
‑
羟基
‑4‑
正辛氧基二苯甲酮、10g双(1,2,2,6,6
‑
五甲基
‑4‑
哌啶基)癸二酸酯、10gβ
‑
(3,5
‑
二叔丁基
‑4‑
羟基苯基)丙酸十八碳醇酯和5g季戊四醇四(3
‑
月桂基硫代丙酸酯)混合均匀,再称取
5kg ka
‑
31(新加坡tpc)颗粒并加入上面混合助剂,机械搅拌30min得到中间层上转换纳米陶瓷eva膜功能预备料。
162.s3:上层反射组合物,即纳米银线复合纳米陶瓷eva膜功能预备料的制备
163.s31:纳米银线复合纳米陶瓷粉体的制备
164.在500ml烧杯中加入7.5g聚乙烯吡咯烷酮,向其中加入200ml无水乙醇,机械搅拌直至聚乙烯吡咯烷酮完全溶解于溶剂中,称取12.5g的纳米银线(线径为15nm,线长为35μm)加入其中,于76℃机械搅拌并冷却回流3小时,然后向其中缓慢加入25g纳米陶瓷ito粉体(sn:in原子比为1:10,d90为30nm),加完后继续搅拌并冷却回流8h,于50℃减压蒸馏回收溶剂,80℃真空干燥12小时得到纳米银线复合纳米陶瓷粉体。
165.s32:纳米银线复合纳米陶瓷分散液的制备
166.称取5g的efka4310分散剂于500ml烧杯中,向其中加入65g无水乙醇和65g异丙醇,搅拌均匀直至分散剂完全溶解于溶剂中,再向混合液中加入15g纳米银线复合纳米陶瓷ito粉体,搅拌均匀,然后超声波分散30min,得到悬浊液。最后将悬浊液转入0.3l涡轮式纳米砂磨机,并加入0.3mm镐珠,控制物料温度25~40℃,转速为2000r/min砂磨时间为3小时得到纳米银线复合纳米陶瓷分散液。
167.s33:上层纳米银线复合纳米陶瓷eva膜功能预备料的制备
168.称取75g的纳米银线复合纳米陶瓷分散液,向其中加入50g 1,1
‑
二叔丁基过氧化
‑
3,3,5
‑
甲基环己烷交联剂、50gγ
‑
甲基丙烯酰氧基丙基三甲氧基硅烷、20g 2
‑
羟基
‑4‑
正辛氧基二苯甲酮、10g双(1,2,2,6,6
‑
五甲基
‑4‑
哌啶基)癸二酸酯、10gβ
‑
(3,5
‑
二叔丁基
‑4‑
羟基苯基)丙酸十八碳醇酯和5g季戊四醇四(3
‑
月桂基硫代丙酸酯)混合均匀,再称取5kg ka
‑
40(新加坡tpc)颗粒并加入上面混合助剂,机械搅拌15min得到上层纳米银线复合纳米陶瓷eva膜功能预备料。
169.s4:三层共挤智能调光eva膜的制备
170.将下层热致变色eva膜功能预备料、中间层上转换纳米陶瓷eva膜功能预备料和上层纳米银线复合纳米陶瓷eva膜功能预备料分别放在流延挤出机的三个进料斗中,控制三条螺杆机筒各温区的温度50~75℃,弯头及连接采用水循环冷却,温度控制为45~60℃,换网器温度控制为48~60℃,模头各温区控制为70~85℃,冷冻机胶辊温度控制为10~15℃,冷冻机铁辊温度控制为20~25℃,通过分配器,流延、挤出、冷却、成型,生产出一种具有三层结构的eva薄膜,包括下层的热致变色eva膜,中间层的上转换纳米陶瓷eva膜以及上层的纳米银线复合纳米陶瓷eva膜,每层厚度为0.26mm,总厚度为0.78mm。
171.为了方便测试,将所得三层共挤智能调光eva膜做成夹层玻璃,按照5mm普白玻璃+0.78mm智能调光eva膜+5mm普白玻璃进行合片,然后放入箱式夹胶炉中,控制真空为
‑
0.1mpa,低温60℃保温20min,高温135℃保温45min,冷却制得智能调光eva夹层玻璃,对其进行光学测试。
172.夹层玻璃的uv
‑
vis
‑
nir透射与正反面反射光谱图(uv
‑
vis
‑
nir透射:光线从上层打入;正面反射:光线由上层打入;反面反射:光线由下层打入)如图3所示。从图3可以看出,夹层玻璃对可见光具有很好的透射能力,对780nm以上的红外光的透射率在20%及以下,尤其是对波长1000nm以上的近红外的透射率极低,透射率可达到0,说明该夹层玻璃对红外具有很好的阻隔作用。同时,从正反面反射光谱图可以看出,夹层玻璃的正面和反面对可见光
具有相似的反射能力,其中正面对近红外的反射率明显高于反面的反射率,反映了纳米银线复合纳米陶瓷粉体可有效提高对近红外的反射能力。
173.采用980nm的光线对夹层玻璃进行激发,得到夹层玻璃在2ma、4ma以及980nm下的发射光谱图如图4所示。图4反映,采用980nm的近红外光线对夹层玻璃进行照射后,夹层玻璃可以发出700nm以下的可见光,这是由于智能调光eva膜的中间层中ce、y共掺杂cs
0.33
wo3纳米陶瓷具有上转换的能力,能够将近红外光线转化为可见光。
174.采用808nm的光线对夹层玻璃进行激发,得到夹层玻璃在1.0ma、1.3ma以及808nm下的发射光谱图如图5所示。图5进一步反映了智能调光eva膜的中间层中ce、y共掺杂cs
0.33
wo3纳米陶瓷具有上转换的能力,能够将近红外光线转化为可见光,不过在光强较弱情况下的上转换能力有所降低。
175.实施例2
176.一种三层共挤智能调光eva膜,包括依次层叠的下层、中间层和上层,其中,下层为热致变色eva膜,中间层为上转换纳米陶瓷eva膜,上层为纳米银线复合纳米陶瓷eva膜。三层共挤智能调光eva膜的制备方法包括如下步骤:
177.s1:下层热致变色组合物,即热致变色eva膜功能预备料的制备
178.先称取25g柠檬酸螯合铜放在500ml烧杯中,向其中加入10g聚乙烯吡咯烷酮,再滴加215g的双环戊二烯乙氧基甲基丙烯酸酯,搅拌均匀后利用超声波细胞粉碎仪超声分散30min,超声波频率为2000hz,得到柠檬酸螯合铜分散液。再将125g peg(分子量为1250)逐滴滴加到柠檬酸螯合铜分散液中,边滴加边搅拌均匀使柠檬酸螯合铜表面完全包覆peg,得到柠檬酸螯合铜/peg的分散液。
179.称取5kg ka
‑
40(新加坡tpc)eva颗粒,将eva在
‑
40℃下冷冻粉碎至350目,然后称取250g柠檬酸螯合铜/peg的分散液、40g 1,1
‑
二特戊基过氧化环己烷、40gγ
‑
甲基丙烯酰氧基丙基三甲氧基硅烷、10g 2
‑
羟基
‑4‑
正辛氧基二苯甲酮和10g纳米氧化锌(d90为10~30nm)、10g双(1,2,2,6,6
‑
五甲基
‑4‑
哌啶基)癸二酸酯、5gβ
‑
(3,5
‑
二叔丁基
‑4‑
羟基苯基)丙酸十八碳醇酯和2.5g季戊四醇四(3
‑
月桂基硫代丙酸酯)混合均匀加入到eva粉末中搅拌均匀并干燥,干燥温度为40℃,搅拌时间为25min,得到热致变色eva膜功能预备料。
180.s2:中间层上转换组合物,即上转换纳米陶瓷eva膜功能预备料的制备
181.s21:ce、y共掺杂cs
0.33
wo3纳米陶瓷(ce
0.01
y
0.01
cs
0.33
wo3)粉体制备
182.称取39.65g(0.1mol)六氯化钨于500ml烧杯中,加200ml无水乙醇溶解得到a液;称取5.56g(0.033mol)氯化铯于50ml烧杯中,加30ml无水乙醇溶解得到b液;称取0.434g(0.001mol)六水合硝酸铈于50ml烧杯中,加20ml无水乙醇溶解得到c液;称取0.383g(0.001mol)六水合硝酸钇于50ml烧杯中,加20ml无水乙醇溶解得到d液。
183.然后将a液转入到500ml四口烧瓶中,于76℃回流搅拌3小时,继续回流搅拌并将b液、c液以及d液分别转入到50ml恒压滴液漏斗中,先将b液以3滴/秒速度缓慢滴加到a液中,待b液滴加完毕后,同时滴加c液和d液并保持速度为2滴/秒,待c液、d液滴加完毕后继续加热回流搅拌3小时,然后冷却至室温并静置陈化24小时得到溶胶。
184.将溶胶减压蒸馏直到得到透明湿凝胶,干燥并粉碎后得到前驱体粉末。再将前驱体粉末在马弗炉中500℃煅烧5h,并通入氮气和氢气混合气体,氮气流量为0.8l/min,氢气流量为0.4l/min,最终得到ce、y共掺杂cs
0.33
wo3纳米陶瓷粉体,研磨称重22.08g。ce、y共掺
杂cs
0.33
wo3纳米陶瓷粉体的结构、形貌与实施例1的相同。
185.s22:ce、y共掺杂cs
0.33
wo3纳米陶瓷分散液的制备
186.称取10g sago9311分散剂于250ml烧杯中,向其中加入170g乙酸乙酯,搅拌均匀直至分散剂完全溶解于溶剂中,再向混合液中加入20g ce、y共掺杂cs
0.33
wo3纳米陶瓷粉体,搅拌均匀,然后超声波分散10min,最后将悬浊液转入0.3l棒硝式纳米砂磨机,并加入0.2mm镐珠,控制物料温度25~40℃,转速为2800r/min,砂磨时间为5小时得到ce、y共掺杂cs
0.33
wo3纳米陶瓷分散液。
187.s23:中间层上转换纳米陶瓷膜eva功能预备料的制备
188.称取125g的ce、y共掺杂cs
0.33
wo3纳米陶瓷分散液,向其中加入40g 1,1
‑
二特戊基过氧化环己烷、40gγ
‑
甲基丙烯酰氧基丙基三甲氧基硅烷、10g 2
‑
羟基
‑4‑
正辛氧基二苯甲酮和10g纳米氧化锌(d90为10~30nm)、10g双(1,2,2,6,6
‑
五甲基
‑4‑
哌啶基)癸二酸酯、5gβ
‑
(3,5
‑
二叔丁基
‑4‑
羟基苯基)丙酸十八碳醇酯和2.5g季戊四醇四(3
‑
月桂基硫代丙酸酯)混合均匀,再称取5kg eva
‑
260(日本三井)颗粒并加入上面混合助剂,机械搅拌30min得到中间层上转换纳米陶瓷eva膜功能预备料。
189.s3:上层反射组合物,即纳米银线复合纳米陶瓷eva膜功能预备料的制备:
190.s31:纳米银线复合纳米陶瓷粉体的制备
191.在500ml烧杯中加入8g kh
‑
570(γ
‑
甲基丙烯酰氧基丙基三甲氧基硅烷),向其中加入200ml无水乙醇,机械搅拌直至kh
‑
570完全溶解于溶剂中,称取10g的纳米银线(线径为15nm,线长为35μm)加入其中,于76℃机械搅拌并冷却回流3小时,然后向其中缓慢加入30g纳米陶瓷ito粉体(sn:in原子比为1:10,d90为25nm),加完后继续搅拌并冷却回流8h,于50℃减压蒸馏回收溶剂,80℃真空干燥12小时得到纳米银线复合纳米陶瓷粉体。
192.s32:纳米银线复合纳米陶瓷分散液的制备
193.称取5g的sago
‑
9105分散剂于500ml烧杯中,向其中加入65g无水乙醇和65g异丙醇,搅拌均匀直至分散剂完全溶解于溶剂中,再向混合液中加入15g纳米银线复合纳米陶瓷ito粉体,搅拌均匀,然后超声波分散30min,最后将悬浊液转入0.3l涡轮式纳米砂磨机,并加入0.3mm镐珠,控制物料温度25~40℃,转速为2000r/min砂磨时间为3小时得到纳米银线复合纳米陶瓷分散液。
194.s33:上层纳米银线复合纳米陶瓷eva膜功能预备料的制备
195.称取75g的纳米银线复合纳米陶瓷分散液,向其中加入40g 1,1
‑
二特戊基过氧化环己烷、40gγ
‑
甲基丙烯酰氧基丙基三甲氧基硅烷、10g 2
‑
羟基
‑4‑
正辛氧基二苯甲酮和10g纳米氧化锌(d90为10~30nm)、10g双(1,2,2,6,6
‑
五甲基
‑4‑
哌啶基)癸二酸酯、5gβ
‑
(3,5
‑
二叔丁基
‑4‑
羟基苯基)丙酸十八碳醇酯和2.5g季戊四醇四(3
‑
月桂基硫代丙酸酯)混合均匀,再称取5kg ka
‑
40(新加坡tpc)颗粒并加入上面混合助剂,机械搅拌15min得到上层纳米银线复合纳米陶瓷eva膜功能预备料。
196.s4:三层共挤智能调光eva膜的制备
197.将下层热致变色eva膜功能预备料、中间层上转换纳米陶瓷eva膜功能预备料和上层纳米银线复合纳米陶瓷eva膜功能预备料分别放在流延挤出机的三个进料斗中,控制三条螺杆机筒各温区的温度50~75℃,弯头及连接采用水循环冷却,温度控制为45~60℃,换网器温度控制为48~60℃,模头各温区控制为70~85℃,冷冻机胶辊温度控制为10~15℃,
冷冻机铁辊温度控制为20~25℃,通过分配器,流延、挤出、冷却、成型,生产出一种具有三层结构的eva薄膜,包括下层的热致变色eva膜,中间层的上转换纳米陶瓷eva膜以及上层的纳米银线复合纳米陶瓷eva膜,每层厚度为0.26mm,总厚度为0.78mm。
198.为了方便测试,将所得三层共挤智能调光eva膜做成夹层玻璃,按照5mm普白玻璃+0.78mm智能调光eva膜+5mm普白玻璃进行合片,然后放入箱式夹胶炉中,控制真空为
‑
0.1mpa,低温60℃保温20min,高温135℃保温45min,冷却制得智能调光eva夹层玻璃,对其进行光学测试。
199.实施例3
200.一种三层共挤智能调光eva膜,包括依次层叠的下层、中间层和上层,其中,下层为热致变色eva膜,中间层为上转换纳米陶瓷eva膜,上层为纳米银线复合纳米陶瓷eva膜。三层共挤智能调光eva膜的制备方法包括如下步骤:
201.s1:下层热致变色组合物,即热致变色eva膜功能预备料的制备
202.称取10g柠檬酸螯合铜放在500ml烧杯中,向其中加入5g聚乙烯吡咯烷酮,再滴加185g的三烯丙基异三聚氰酸酯(taic),搅拌均匀后利用超声波细胞粉碎仪超声分散30min,超声波频率为2000hz,得到柠檬酸螯合铜分散液。再将100g peg(分子量为1250)逐滴滴加到柠檬酸螯合铜分散液中,边滴加边搅拌均匀使柠檬酸螯合铜表面完全包覆peg,得到柠檬酸螯合铜/peg的分散液。
203.称取5kg 282pv(韩国韩华)eva颗粒,将eva在
‑
40℃下冷冻粉碎至200目,然后称取300g柠檬酸螯合铜/peg的分散液、50g 2,5
‑
二甲基
‑
2,5
‑
二(2
‑
乙基己酰过氧化)己烷、50gγ
‑
甲基丙烯酰氧基丙基三甲氧基硅烷、20g 2
‑
(2'
‑
羟基
‑
5'
‑
特辛基)苯基苯并三氮唑、10g双(1,2,2,6,6
‑
五甲基
‑4‑
哌啶基)癸二酸酯、5gβ
‑
(3,5
‑
二叔丁基
‑4‑
羟基苯基)丙酸十八碳醇酯和2.5g季戊四醇四(3
‑
月桂基硫代丙酸酯)混合均匀加入到eva粉末中搅拌均匀并干燥,干燥温度为40℃,搅拌时间为25min,得到热致变色eva膜功能预备料。
204.s2:中间层上转换组合物,即上转换纳米陶瓷eva膜功能预备料的制备
205.s21:ce、y共掺杂cs
0.33
wo3纳米陶瓷(ce
0.03
y
0.03
cs
0.33
wo3)粉体制备
206.称取39.65g(0.1mol)六氯化钨于500ml烧杯中,加200ml无水乙醇溶解得到a液;称取5.56g(0.033mol)氯化铯于50ml烧杯中,加30ml无水乙醇溶解得到b液;称取1.30g(0.003mol)六水合硝酸铈于50ml烧杯中,加20ml无水乙醇溶解得到c液;称取1.15g(0.003mol)六水合硝酸钇于50ml烧杯中,加20ml无水乙醇溶解得到d液。
207.然后将a液转入到500ml四口烧瓶中,于76℃回流搅拌3小时,继续回流搅拌并将b液、c液以及d液分别转入到50ml恒压滴液漏斗中,先将b液以3滴/秒速度缓慢滴加到a液中,待b液滴加完毕后,同时滴加c液和d液并保持速度为2滴/秒,待c液、d液滴加完毕后继续加热回流搅拌3小时,然后冷却至室温并静置陈化24小时得到溶胶。
208.将溶胶减压蒸馏直到得到透明湿凝胶,干燥并粉碎后得到前驱体粉末,再将前驱体粉末在马弗炉中500℃煅烧5h,并通入氮气和氢气混合气体,氮气流量为0.8l/min,氢气流量为0.4l/min,最终得到上转换ce、y共掺杂cs
0.33
wo3纳米陶瓷粉体,研磨称重22.58g。
209.s22:ce、y共掺杂cs
0.33
wo3纳米陶瓷分散液的制备
210.称取10g efka4310分散剂于500ml烧杯中,向其中加入170g丙二醇甲醚醋酸酯,搅拌均匀直至分散剂完全溶解于溶剂中,再向混合液中加入20g ce、y共掺杂cs
0.33
wo3纳米陶
瓷粉体,搅拌均匀,然后超声波分散10min,最后将悬浊液转入0.3l棒硝式纳米砂磨机,并加入0.2mm镐珠,控制物料温度25~40℃,转速为2800r/min,砂磨时间为5小时得到ce、y共掺杂cs
0.33
wo3纳米陶瓷分散液。
211.s23:中间层上转换纳米陶瓷eva膜功能预备料的制备
212.称取125g的ce、y共掺杂cs
0.33
wo3纳米陶瓷分散液,向其中加入50g 2,5
‑
二甲基
‑
2,5
‑
二(2
‑
乙基己酰过氧化)己烷、50gγ
‑
甲基丙烯酰氧基丙基三甲氧基硅烷、20g 2
‑
(2'
‑
羟基
‑
5'
‑
特辛基)苯基苯并三氮唑、10g双(1,2,2,6,6
‑
五甲基
‑4‑
哌啶基)癸二酸酯、5gβ
‑
(3,5
‑
二叔丁基
‑4‑
羟基苯基)丙酸十八碳醇酯和2.5g季戊四醇四(3
‑
月桂基硫代丙酸酯)混合均匀,再称取5kg 28005(韩国lg)颗粒并加入上面混合助剂,机械搅拌30min得到中间层上转换纳米陶瓷eva膜功能预备料。
213.s3:上层反射组合物,即纳米银线复合纳米陶瓷eva膜功能预备料的制备
214.s31:纳米银线复合纳米陶瓷粉体的制备
215.在500ml烧杯中加入7g聚乙烯吡咯烷酮,向其中加入200ml无水乙醇,机械搅拌直至聚乙烯吡咯烷酮完全溶解于溶剂中,称取5g的纳米银线(线径为15nm,线长为35μm)加入其中,于76℃机械搅拌并冷却回流3小时,然后向其中缓慢加入30g纳米陶瓷ito粉体(sn:in原子比为1:10,d90为15nm),加完后继续搅拌并冷却回流8h,于50℃减压蒸馏回收溶剂,80℃真空干燥12小时得到纳米银线复合纳米陶瓷粉体。
216.s32:纳米银线复合纳米陶瓷分散液的制备
217.称取6g的byk
‑
2200于500ml烧杯中,向其中加入150g无水乙醇和79g异丙醇,搅拌均匀直至分散剂完全溶解于溶剂中,再向混合液中加入15g纳米银线复合纳米陶瓷ito粉体,搅拌均匀,然后超声波分散30min,最后将悬浊液转入0.3l涡轮式纳米砂磨机,并加入0.3mm镐珠,控制物料温度25~40℃,转速为2000r/min砂磨时间为3小时得到纳米银线复合纳米陶瓷分散液。
218.s33:上层纳米银线复合纳米陶瓷eva膜功能预备料的制备
219.称取75g的纳米银线复合纳米陶瓷分散液,向其中加入50g 2,5
‑
二甲基
‑
2,5
‑
二(2
‑
乙基己酰过氧化)己烷、50gγ
‑
甲基丙烯酰氧基丙基三甲氧基硅烷、20g 2
‑
(2'
‑
羟基
‑
5'
‑
特辛基)苯基苯并三氮唑、10g双(1,2,2,6,6
‑
五甲基
‑4‑
哌啶基)癸二酸酯、5gβ
‑
(3,5
‑
二叔丁基
‑4‑
羟基苯基)丙酸十八碳醇酯和2.5g季戊四醇四(3
‑
月桂基硫代丙酸酯)混合均匀,再称取5kg eva
‑
150(日本三井)颗粒并加入上面混合助剂,机械搅拌15min得到上层纳米银线复合纳米陶瓷eva膜功能预备料。
220.s4:三层共挤智能调光eva膜的制备
221.将下层热致变色eva膜功能预备料、中间层上转换纳米陶瓷eva膜功能预备料和上层纳米银线复合纳米陶瓷eva膜功能预备料分别放在流延挤出机的三个进料斗中,控制三条螺杆机筒各温区的温度50~75℃,弯头及连接采用水循环冷却,温度控制为45~60℃,换网器温度控制为48~60℃,模头各温区控制为70~85℃,冷冻机胶辊温度控制为10~15℃,冷冻机铁辊温度控制为20~25℃,通过分配器,流延、挤出、冷却、成型,生产出一种具有三层结构的eva薄膜,包括下层的热致变色eva膜,中间层的上转换纳米陶瓷eva膜以及上层的纳米银线复合纳米陶瓷eva膜,每层厚度为0.26mm,总厚度为0.78mm。
222.为了方便测试,将所得样品做成夹层玻璃,按照5mm普白玻璃+0.78mm智能调光eva
膜+5mm普白玻璃进行合片,然后放入箱式夹胶炉中,控制真空为
‑
0.1mpa,低温60℃保温20min,高温135℃保温45min,冷却制得智能调光eva夹层玻璃,对其进行光学测试。
223.实施例4
224.一种三层共挤智能调光eva膜,包括依次层叠的下层、中间层和上层,其中,下层为热致变色eva膜,中间层为上转换纳米陶瓷eva膜,上层为纳米银线复合纳米陶瓷eva膜。三层共挤智能调光eva膜的制备方法包括如下步骤:
225.s1:下层热致变色组合物,即热致变色eva膜功能预备料的制备
226.称取3g柠檬酸螯合铜放在500ml烧杯中,向其中加入1g聚乙烯吡咯烷酮,再滴加296g的三羟甲基丙烷三甲基丙烯酸酯(tmptma),搅拌均匀后利用超声波细胞粉碎仪超声分散10min,超声波频率为2000hz,得到柠檬酸螯合铜分散液。再将60g peg(分子量为1250)逐滴滴加到柠檬酸螯合铜分散液中,边滴加边搅拌均匀使柠檬酸螯合铜表面完全包覆peg,得到柠檬酸螯合铜/peg的分散液。
227.称取5kg ka
‑
40(新加坡tpc)eva颗粒,将eva在
‑
40℃下冷冻粉碎至350目,然后称取250g柠檬酸螯合铜/peg的分散液、40g 1,1
‑
二叔丁基过氧化
‑
3,3,5
‑
甲基环己烷、40g氨丙基三甲氧基硅烷、20g 2
‑
(4,6
‑
双(2,4
‑
二甲基苯基)
‑
1,3,5
‑
三嗪
‑2‑
基)
‑5‑
辛氧基酚、10g聚丁二酸(4
‑
羟基
‑
2,2,6,6
‑
四甲基
‑1‑
哌啶乙醇)酯、5gβ
‑
(3,5
‑
二叔丁基
‑4‑
羟基苯基)丙酸十八碳醇酯和2.5g季戊四醇四(3
‑
月桂基硫代丙酸酯)混合均匀加入到eva粉末中搅拌均匀并干燥,干燥温度为40℃,搅拌时间为25min,得到下层热致变色eva膜功能预备料。
228.s2:中间层上转换组合物,即上转换纳米陶瓷eva膜功能预备料的制备
229.s21:ce、y共掺杂cs
0.33
wo3纳米陶瓷(ce
0.001
y
0.001
cs
0.33
wo3)粉体制备
230.称取39.65g(0.1mol)六氯化钨于500ml烧杯中,加200ml无水乙醇溶解得到a液;称取5.56g(0.033mol)氯化铯于50ml烧杯中,加30ml无水乙醇溶解得到b液;称取0.0434g(0.0001mol)六水合硝酸铈于50ml烧杯中,加20ml无水乙醇溶解得到c液;称取0.0383g(0.0001mol)六水合硝酸钇于50ml烧杯中,加20ml无水乙醇溶解得到d液。
231.然后将a液转入到500ml四口烧瓶中,于76℃回流搅拌3小时,继续回流搅拌并将b液、c液以及d液分别转入到50ml恒压滴液漏斗中,先将b液以3滴/秒速度缓慢滴加到a液中,待b液滴加完毕后,同时滴加c液和d液并保持速度为2滴/秒,待c液、d液滴加完毕后继续加热回流搅拌3小时,然后冷却至室温并静置陈化24小时得到溶胶。
232.将溶胶减压蒸馏直到得到透明湿凝胶,干燥并粉碎后得到前驱体粉末,再将前驱体粉末在马弗炉中500℃煅烧5h,并通入氮气和氢气混合气体,氮气流量为0.6l/min,氢气流量为0.3l/min,最终得到上转换ce、y共掺杂cs
0.33
wo3纳米陶瓷粉体,研磨称重21.31g。
233.s22:ce、y共掺杂cs
0.33
wo3纳米陶瓷分散液的制备
234.称取8g sago9311分散剂于250ml烧杯中,向其中加入105g环己烷,搅拌均匀直至分散剂完全溶解于溶剂中,再向混合液中加入20g ce、y共掺杂cs
0.33
wo3纳米陶瓷粉体,搅拌均匀,然后超声波分散10min,最后将悬浊液转入0.3l棒硝式纳米砂磨机,并加入0.2mm镐珠,控制物料温度25~40℃,转速为2500r/min,砂磨时间为5小时得到ce、y共掺杂cs
0.33
wo3纳米陶瓷分散液。
235.s23:中间层上转换纳米陶瓷eva膜功能预备料的制备
236.称取125g的ce、y共掺杂cs
0.33
wo3纳米陶瓷分散液,向其中加入40g 1,1
‑
二叔丁基
过氧化
‑
3,3,5
‑
甲基环己烷、40g氨丙基三甲氧基硅烷、20g 2
‑
(4,6
‑
双(2,4
‑
二甲基苯基)
‑
1,3,5
‑
三嗪
‑2‑
基)
‑5‑
辛氧基酚、10g聚丁二酸(4
‑
羟基
‑
2,2,6,6
‑
四甲基
‑1‑
哌啶乙醇)酯、5gβ
‑
(3,5
‑
二叔丁基
‑4‑
羟基苯基)丙酸十八碳醇酯和2.5g季戊四醇四(3
‑
月桂基硫代丙酸酯)混合均匀,再称取5kg mv
‑
1055(泰国)颗粒并加入上面混合助剂,机械搅拌30min得到中间层上转换纳米陶瓷eva膜功能预备料。
237.s3:上层反射组合物,即纳米银线复合纳米陶瓷eva膜功能预备料的制备:
238.s31:纳米银线复合纳米陶瓷粉体的制备
239.在500ml烧杯中加入15g kh
‑
570(γ
‑
甲基丙烯酰氧基丙基三甲氧基硅烷),向其中加入300ml无水乙醇,机械搅拌直至kh
‑
570完全溶解于溶剂中,称取15g的纳米银线(线径为15nm,线长为35μm)加入其中,于76℃机械搅拌并冷却回流3小时,然后向其中缓慢加入60g纳米陶瓷ito粉体(sn:in原子比为1:10,d90为20nm),加完后继续搅拌并冷却回流8h,于50℃减压蒸馏回收溶剂,80℃真空干燥12小时得到纳米银线复合纳米陶瓷粉体。
240.s32:纳米银线复合纳米陶瓷分散液的制备
241.称取10g的sago
‑
9105于500ml烧杯中,向其中加入80g正丁醇和80g异丙醇,搅拌均匀直至分散剂完全溶解于溶剂中,再向混合液中加入30g纳米银线复合纳米陶瓷ito粉体,搅拌均匀,然后超声波分散30min,最后将悬浊液转入0.3l涡轮式纳米砂磨机,并加入0.3mm镐珠,控制物料温度25~40℃,转速为2000r/min砂磨时间为3小时得到纳米银线复合纳米陶瓷分散液。
242.s33:上层纳米银线复合纳米陶瓷eva膜功能预备料的制备
243.称取75g的纳米银线复合纳米陶瓷分散液,向其中加入40g 1,1
‑
二叔丁基过氧化
‑
3,3,5
‑
甲基环己烷、40g氨丙基三甲氧基硅烷、20g 2
‑
(4,6
‑
双(2,4
‑
二甲基苯基)
‑
1,3,5
‑
三嗪
‑2‑
基)
‑5‑
辛氧基酚、10g聚丁二酸(4
‑
羟基
‑
2,2,6,6
‑
四甲基
‑1‑
哌啶乙醇)酯、5gβ
‑
(3,5
‑
二叔丁基
‑4‑
羟基苯基)丙酸十八碳醇酯和2.5g季戊四醇四(3
‑
月桂基硫代丙酸酯)混合均匀,再称取5kg eva
‑
150(日本三井)颗粒并加入上面混合助剂,机械搅拌15min得到上层纳米银线复合纳米陶瓷eva膜功能预备料。
244.s4:三层共挤智能调光eva膜的制备
245.将下层热致变色eva膜功能预备料、中间层上转换纳米陶瓷eva膜功能预备料和上层纳米银线复合纳米陶瓷eva膜功能预备料分别放在流延挤出机的三个进料斗中,控制三条螺杆机筒各温区的温度50~75℃,弯头及连接采用水循环冷却,温度控制为45~60℃,换网器温度控制为48~60℃,模头各温区控制为70~85℃,冷冻机胶辊温度控制为10~15℃,冷冻机铁辊温度控制为20~25℃,通过分配器,流延、挤出、冷却、成型,生产出一种具有三层结构的eva薄膜,包括下层的热致变色eva膜,中间层的上转换纳米陶瓷eva膜以及上层的纳米银线复合纳米陶瓷eva膜,每层厚度为0.26mm,总厚度为0.78mm。
246.为了方便测试,将所得三层共挤智能调光eva膜做成夹层玻璃,按照5mm普白玻璃+0.78mm智能调光eva膜+5mm普白玻璃进行合片,然后放入箱式夹胶炉中,控制真空为
‑
0.1mpa,低温60℃保温20min,高温135℃保温45min,冷却制得智能调光eva夹层玻璃,对其进行光学测试。
247.对比例1
248.本对比例提供一种单层eva膜,由纳米银线复合纳米陶瓷分散液、ce、y共掺杂纳米
陶瓷分散液、檬酸螯合铜/peg的分散液混合在一起,然后添加到eva颗粒中搅拌均匀直接一层流延挤出而得。具体的制备方法为:
249.将250g柠檬酸螯合铜/peg的分散液、80g ce、y共掺杂cs
0.33
wo3纳米陶瓷分散液和75g纳米银线复合纳米陶瓷分散液混合均匀,得到混合分散液。称取5kg日本三井eva
‑
150颗粒,在
‑
40℃下冷冻粉碎至350目,然后称取50g 1,1
‑
二叔丁基过氧化
‑
3,3,5
‑
甲基环己烷、50gγ
‑
甲基丙烯酰氧基丙基三甲氧基硅烷、20g 2
‑
羟基
‑4‑
正辛氧基二苯甲酮、10g双(1,2,2,6,6
‑
五甲基
‑4‑
哌啶基)癸二酸酯、10gβ
‑
(3,5
‑
二叔丁基
‑4‑
羟基苯基)丙酸十八碳醇酯和5g季戊四醇四(3
‑
月桂基硫代丙酸酯)与上面混合分散液混合均匀加入到eva粉末中搅拌均匀并干燥,干燥温度为40℃,搅拌时间为25min,得到eva膜功能预备料。其中,柠檬酸螯合铜/peg的分散液、ce、y共掺杂cs
0.33
wo3纳米陶瓷分散液和纳米银线复合纳米陶瓷分散液的制备方法与实施例1相同。
250.将eva膜功能预备料放在流延挤出机的一个进料斗中,采用单螺杆挤出机挤出,控制螺杆机筒各温区的温度50~75℃,弯头及连接采用水循环冷却,温度控制为45~60℃,换网器温度控制为48~60℃,模头各温区控制为70~85℃,冷冻机胶辊温度控制为10~15℃,冷冻机铁辊温度控制为20~25℃,经流延、挤出、冷却、成型,控制厚度为0.78mm,生产出一种具有单层结构的eva薄膜。
251.为了方便测试,将所得eva薄膜做成夹层玻璃,按照5mm普白玻璃+0.78mm eva膜+5mm普白玻璃进行合片,然后放入箱式夹胶炉中,控制真空为
‑
0.1mpa,低温60℃保温20min,高温135℃保温45min,冷却制得eva夹层玻璃,对其进行光学测试。
252.对比例2
253.本对比例提供一种三层共挤智能调光eva膜,与实施例1的区别在于:本对比例的上层纳米银线复合纳米陶瓷eva膜功能预备料中没有加入纳米银线。其他操作与实施例1相同。
254.对比例3
255.本对比例提供一种三层共挤智能调光eva膜,与实施例1的区别在于:本对比例的纳米银线复合纳米陶瓷粉体中,纳米银线的加入量为2.5g(步骤s31,使ag与ito质量比为1:10)。其他操作与实施例1相同。
256.对比例4
257.本对比例提供一种三层共挤智能调光eva膜,与实施例1的区别在于:本对比例将上层纳米银线复合纳米陶瓷eva膜功能预备料中的纳米银线替换为等质量的纳米银颗粒(粒径≤20nm,步骤s31)。其他操作与实施例1相同。
258.对比例5
259.本对比例提供一种三层共挤智能调光eva膜,与实施例1的区别在于:本对比例中,上层纳米银线复合纳米陶瓷eva膜功能预备料的纳米银线复合纳米陶瓷分散液用量为125g(使纳米银线复合纳米陶瓷分散液为eva颗粒的2.5wt%,步骤s33)。其他操作与实施例1相同。
260.对比例6
261.本对比例提供一种三层共挤智能调光eva膜,与实施例1的区别在于:在制备ce、y共掺杂cs
0.33
wo3纳米陶瓷粉体步骤(步骤s21)中,增加ce、y的掺杂量。具体地,将六水合硝酸
铈和六水合硝酸钇的用量分别改为4.34g(0.01mol)和1.92g(0.005mol)。其他操作与实施例1相同。
262.对比例7
263.本对比例提供一种三层共挤智能调光eva膜,与实施例1的区别在于:在制备ce、y共掺杂cs
0.33
wo3纳米陶瓷粉体步骤(步骤s21)中,ce、y的掺杂量为0,即没有加入六水合硝酸铈和六水合硝酸钇。其他操作与实施例1相同。
264.对比例8
265.本对比例提供一种三层共挤智能调光eva膜,与实施例1的区别在于:将步骤s21中的ce、y共掺杂cs
0.33
wo3纳米陶瓷粉体替换为ce、bi共掺杂cs
0.33
wo3纳米陶瓷粉体,各元素的摩尔比为:ce:bi:cs:w=0.01:0.04:0.33:1。ce、bi共掺杂cs
0.33
wo3纳米陶瓷粉体的制备方法参考ce、y共掺杂cs
0.33
wo3纳米陶瓷粉体。
266.对比例9
267.本对比例提供一种三层共挤智能调光eva膜,与实施例1的区别在于:将步骤s21中的ce、y共掺杂cs
0.33
wo3纳米陶瓷粉体替换为ce、sb共掺杂cs
0.33
wo3纳米陶瓷粉体,各元素的摩尔比为:ce:sb:cs:w=0.05:0.05:0.33:1。ce、sb共掺杂cs
0.33
wo3纳米陶瓷粉体的制备方法参考ce、y共掺杂cs
0.33
wo3纳米陶瓷粉体。
268.对比例10
269.本对比例提供一种三层共挤智能调光eva膜,与实施例1的区别在于:将步骤s21中的ce、y共掺杂cs
0.33
wo3纳米陶瓷粉体替换为y、sb共掺杂cs
0.33
wo3纳米陶瓷粉体,各元素的摩尔比为:y:sb:cs:w=0.02:0.03:0.33:1。y、sb共掺杂cs
0.33
wo3纳米陶瓷粉体的制备方法参考ce、y共掺杂cs
0.33
wo3纳米陶瓷粉体。
270.对比例11
271.本对比例提供一种三层共挤智能调光eva膜,与实施例1的区别在于:将步骤s21中的ce、y共掺杂cs
0.33
wo3纳米陶瓷粉体替换为y、bi共掺杂cs
0.33
wo3纳米陶瓷粉体,各元素的摩尔比为:y:bi:cs:w=0.001:0.1:0.33:1。y、bi共掺杂cs
0.33
wo3纳米陶瓷粉体的制备方法参考ce、y共掺杂cs
0.33
wo3纳米陶瓷粉体。
272.对比例12
273.本对比例提供一种三层共挤智能调光eva膜,与实施例1的区别在于:将步骤s23中ce、y共掺杂cs
0.33
wo3纳米陶瓷分散液的用量改为150g,使其掺量为eva颗粒的3wt%。其他操作与实施例1相同。
274.对比例13
275.本对比例提供一种三层共挤智能调光eva膜,与实施例1的区别在于:在步骤s1中,柠檬酸螯合铜的质量为2.5g,使peg与柠檬酸螯合铜质量比为50:1。其他操作与实施例1相同。
276.对比例14
277.本对比例提供一种三层共挤智能调光eva膜,与实施例1的区别在于:在步骤s1中,柠檬酸螯合铜的质量为62.5g,使peg与柠檬酸螯合铜质量比为2:1。其他操作与实施例1相同。
278.对比例15
279.本对比例提供一种三层共挤智能调光eva膜,与实施例1的区别在于:在步骤s1中,peg的分子量为5000。其他操作与实施例1相同。
280.对比例16
281.本对比例提供一种三层共挤智能调光eva膜,与实施例1的区别在于:eva
‑
150颗粒不作冷冻粉碎处理,直接使用。其他操作与实施例1相同。
282.对比例17
283.本对比例提供一种三层共挤智能调光eva膜,与实施例1的区别在于:在步骤s1中,柠檬酸螯合铜/peg的分散液的用量为750g,使其掺量为eva颗粒的15wt%。其他操作与实施例1相同。
284.对比例18
285.本对比例提供一种三层共挤智能调光eva膜,与实施例1的区别在于:下层没有添加柠檬酸螯合铜/peg的分散液,即,将热致变色eva膜功能预备料改为普通的eva膜预备料用于制作下层;且eva不经过冷冻粉碎处理。具体地,下层eva膜预备料的制备方法为:称取5kg日本三井eva
‑
150颗粒,然后称取50g 1,1
‑
二叔丁基过氧化
‑
3,3,5
‑
甲基环己烷、50gγ
‑
甲基丙烯酰氧基丙基三甲氧基硅烷、20g 2
‑
羟基
‑4‑
正辛氧基二苯甲酮、10g双(1,2,2,6,6
‑
五甲基
‑4–
哌啶基)癸二酸酯、10gβ
‑
(3,5
‑
二叔丁基
‑4‑
羟基苯基)丙酸十八碳醇酯和5g季戊四醇四(3
‑
月桂基硫代丙酸酯)混合均匀加入到eva颗粒中搅拌均匀并干燥,干燥温度为40℃,搅拌时间为25min,得到eva膜预备料。
286.其他操作与实施例1相同。
287.对比例19
288.本对比例提供一种三层共挤智能调光eva膜,与实施例1的区别在于:中间层没有添加ce、y共掺杂cs
0.33
wo3纳米陶瓷分散液,即,将上转换纳米陶瓷eva膜功能预备料改为普通的eva膜预备料用于制作中间层。具体地,中间层eva膜预备料的制备方法为:称取50g 1,1
‑
二叔丁基过氧化
‑
3,3,5
‑
甲基环己烷交联剂、50gγ
‑
甲基丙烯酰氧基丙基三甲氧基硅烷、20g 2
‑
羟基
‑4‑
正辛氧基二苯甲酮、10g双(1,2,2,6,6
‑
五甲基
‑4–
哌啶基)癸二酸酯、10gβ
‑
(3,5
‑
二叔丁基
‑4‑
羟基苯基)丙酸十八碳醇酯和5g季戊四醇四(3
‑
月桂基硫代丙酸酯)混合均匀,再称取5kg ka
‑
31(新加坡tpc)颗粒并加入上面混合助剂,机械搅拌30min得到eva膜预备料。
289.其他操作与实施例1相同。
290.对比例20
291.本对比例提供一种三层共挤智能调光eva膜,与实施例1的区别在于:上层没有添加纳米银线复合纳米陶瓷分散液,即,将纳米银线复合纳米陶瓷eva膜功能预备料改为普通的eva膜预备料用于制作上层。具体地,上层eva膜预备料的制备方法为:称取50g 1,1
‑
二叔丁基过氧化
‑
3,3,5
‑
甲基环己烷交联剂、50gγ
‑
甲基丙烯酰氧基丙基三甲氧基硅烷、20g 2
‑
羟基
‑4‑
正辛氧基二苯甲酮、10g双(1,2,2,6,6
‑
五甲基
‑4–
哌啶基)癸二酸酯、10gβ
‑
(3,5
‑
二叔丁基
‑4‑
羟基苯基)丙酸十八碳醇酯和5g季戊四醇四(3
‑
月桂基硫代丙酸酯)混合均匀,再称取5kg ka
‑
40(新加坡tpc)颗粒并加入上面混合助剂,机械搅拌15min得到eva膜预备料。
292.其他操作与实施例1相同。
293.对比例21
294.本对比例提供一种三层共挤智能调光eva膜,与实施例1的区别在于:在制备层热致变色eva膜功能预备料、中间层上转换纳米陶瓷eva膜功能预备料和上层纳米银线复合纳米陶瓷eva膜功能预备料所采用的eva原料全部为日本三井eva
‑
150。
295.实施例1~4以及对比例1~21的光学性能测试以及传热性能测试结果如下表1和表2所示。
296.表1.性能测试结果
[0297][0298][0299]
表2.性能测试结果
[0300][0301]
根据表1和表2的测试结果可以发现:
[0302]
一、对比例1反映,如果把所有分散液混在一起,采用单一膜层流延成膜,容易造成各层功能材料的光谱选择性吸收、透射、反射性能无法充分反映出来,并且存在彼此之间的兼容性问题,最终导致雾度也偏大;
[0303]
二、对比例2由于没有纳米银线的近红外反射,导致对800~2500nm反射率明显降低;
[0304]
三、对比例3由于ito的比例过大,容易阻挡纳米银线的近红外反射,导致对800~2500nm反射率也发生降低;
[0305]
四、对比例4中采用纳米颗粒无法对近红外产生有效的反射,通过与实施例1比较反映出,线状的纳米银线相对于纳米银颗粒能够对近红外进行更充分的反射;
[0306]
五、对比例5中纳米银线复合纳米陶瓷ito分散液添加量偏大,容易造成纳米材料在eva中的团聚,最终导致性能不佳,雾度也偏大;
[0307]
六、对比例6中,ce、y的掺杂量偏大,容易造成纳米晶粒发生晶格畸变,缺陷增多,散射增大,透光率变低;
[0308]
七、对比例7~11反映,当中间层采用不掺杂,或采用稀土与过渡金属对cs
0.33
wo3进行共掺杂时,会引起550~700nm增透率的明显降低。出现这种现象主要是由于稀土、过渡金属掺杂采用的是替位掺杂,虽然可以使晶粒更细,但掺杂量过大,容易造成晶格畸变,甚至
出现杂相,影响纳米材料的综合性能,无法表现出上转换功能。而实施例1采用ce、y共掺杂后,可以将近红外线(808nm、980nm)转换为红橙光(600nm~750nm),从而增强可见光的透过;
[0309]
八、对比例12反映,当ce、y共掺杂纳米陶瓷分散液添加量过多时,容易造成纳米材料在eva中的团聚,进而影响材料综合性能,同时造成雾度偏大;
[0310]
九、对比例13反映,当peg量过大时,造成
‑
oh:铜离子的比例偏大,使得氢氧化铜凝胶簇相对偏少,从而造成变色后的对比度不高;
[0311]
十、对比例14反映,当柠檬酸螯合铜相对peg量偏大时,羟基提供量偏少,这样造成凝胶网不够致密,从而造成变色后的对比度不高;
[0312]
十一、对比例15中,由于peg分子量偏大时,此时的peg已经处于固态状,需要大量的光交联溶剂溶解,并且粘度偏大,阻止了铜离子的水解和醇解,进而造成氢氧化铜凝胶簇相对偏少,导致变色后的对比度不高;
[0313]
十二、对比例16中,当下层的eva不作冷冻粉碎处理,由于柠檬酸螯合铜/peg分散液的添加量偏大,并且系统的极性也偏大,颗粒状的eva很难将分散液充分吸收,最终导致变色后的对比度降低,雾度偏高;而实施例1将eva冷冻粉碎后的接触面积更大,再辅助搅拌并加热,使分散液很好地黏附在eva表面;
[0314]
十三、对比例17中,由于柠檬酸螯合铜/peg的分散液添加量过大,以致分散液很难被粉碎后的eva进一步吸收,加热搅拌时间越长,柠檬酸螯合铜逐渐分离出来,雾度偏大;
[0315]
十四、对比例18的下层省去柠檬酸螯合铜/peg,使得对比度降为零;
[0316]
十五、对比例19的中间层去掉ce、y共掺杂cs
0.33
wo3纳米陶瓷材料,结果不能增强可见光的透过;
[0317]
十六、对比例20的上层去掉纳米银线复合纳米陶瓷材料后,800~2500nm的近红外反射变小;
[0318]
十七、对比例21的上层、中间层、下层的eva原料全部换成日本三井eva
‑
150后,由于三层材料流动性一致,当eva料经过分配器出来后,容易形成三层膜彼此之间的渗透,功能材料彼此之间混在一起,造成材料综合性能受损,同时雾度也偏大。
[0319]
上述实施例为本发明较佳的实施方式,但本发明的实施方式并不受上述实施例的限制,其他的任何未背离本发明的精神实质与原理下所作的改变、修饰、替代、组合、简化,均应为等效的置换方式,都包含在本发明的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1