S-(-)-α-甲基苄胺手性碳量子点的制备方法及应用
本发明属于手性识别和检测,具体涉及s-(-)-α-甲基苄胺手性碳量子点的制备方法及应用。
背景技术:
1、手性碳量子点是指将手性引入到碳量子点中,并影响碳量子点的潜在应用。一般来说,合成手性碳量子点的方法与碳量子点基本上一致,有两种广泛而全面的方法:“自上而下”法和“自下而上”法。前者的制备方法多为化学氧化、激光处理和电化学方法等,用到的反应前体多为石墨烯片、碳纳米管等。后者的合成方法多为化学合成,并涉及到反应前体的聚合和碳化。一般来说,“自上而下”的合成方法工艺相对复杂,合成效率低(即大部分产物为混合物),不容易进行手性碳量子点的定向合成。“自下而上”法被更广泛地适用在制备手性碳量子点上,最常见的非共轭分子前体包括柠檬酸与氨、乙醇胺、乙二胺及其衍生物、l-半胱氨酸、尿素和硫脲的组合,共轭分子前体包括苯二胺及其衍生物,苯酚及其衍生物。
2、手性识别一直是一项具有挑战性的工作,光致发光识别具有灵敏度高和操作过程简单等特点,在手性识别中得到越来越多的应用。在过去的几十年中,报道了一系列手性荧光探针包括有机分子、超分子、纳米材料和多孔材料,并应用于手性识别和检测。碳量子点在立体特异性分子识别中的适用性为手性光学传感器的设计打开了一个新窗口。迄今为止,已经使用手性前体制备了多种具有内在手性的手性碳量子点。
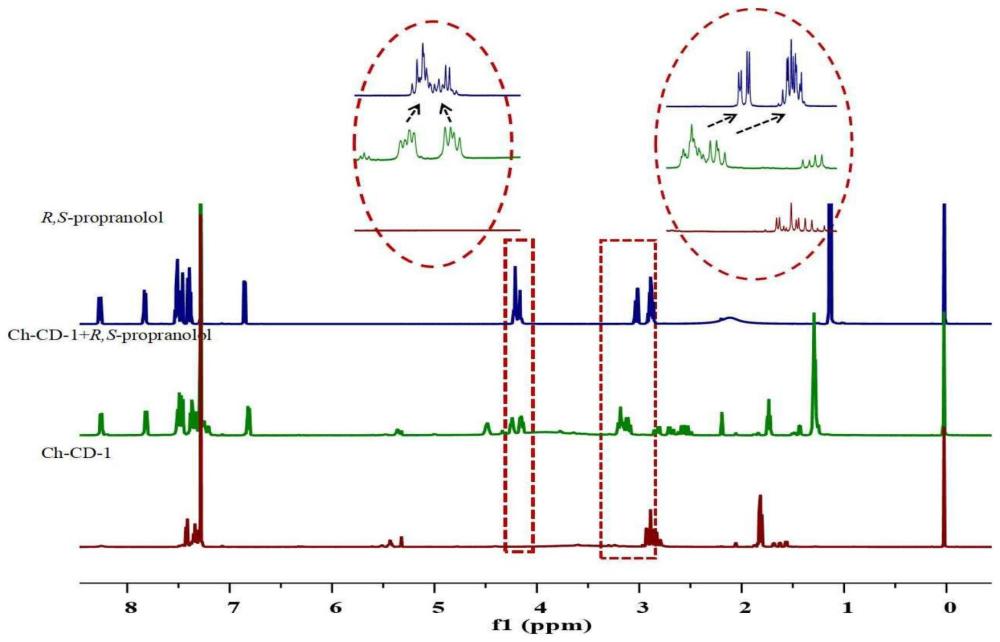
3、目前,基于s-(-)-α-甲基苄胺手性碳量子点用于氨基醇类化合物如普萘洛尔或阿替洛尔对映体的手性识别还未见报道。
技术实现思路
1、本发明旨在解决上述技术问题,提供s-(-)-α-甲基苄胺手性碳量子点的制备方法及应用。
2、本发明的技术方案为:
3、s-(-)-α-甲基苄胺手性碳量子点的制备方法,包括以下步骤:以柠檬酸作为非手性碳源,(s)-(-)-α-甲基苄胺作为手性配体,采用微波辅助一步法制备得到所述s-(-)-α-甲基苄胺手性碳量子点。
4、进一步地,所述的s-(-)-α-甲基苄胺手性碳量子点的制备方法,包括以下步骤:
5、取8-12mmol柠檬酸和1-3mmols-(-)-α-甲基苄胺,加入到8-12ml去离子水中,使二者充分溶解后,将其转移至微波合成仪中,调节功率为400-600w,温度为170-190℃,微波辐射80-120min,待反应完成后,自然冷却至室温,所得溶液经过透析提纯后,得到所述手性碳量子点。
6、优选地,所述的s-(-)-α-甲基苄胺手性碳量子点的制备方法,包括以下步骤:
7、取10mmol柠檬酸和2mmols-(-)-α-甲基苄胺,加入到10ml去离子水中,使二者充分溶解后,将其转移至微波合成仪中,调节功率为500w,温度为180℃,微波辐射100min,待反应完成后,自然冷却至室温,所得溶液经过透析提纯后,得到所述手性碳量子点。
8、本发明还包括上述方法制备得到的s-(-)-α-甲基苄胺手性碳量子点。
9、本发明还提供了上述的s-(-)-α-甲基苄胺手性碳量子点在手性氨基醇类化合物的手性识别中的应用,所述氨基醇类化合物为普萘洛尔或阿替洛尔对映体。
10、本发明的应用包括以下步骤:将普萘洛尔或阿替洛尔对映体与s-(-)-α-甲基苄胺手性碳量子点混合后测定出谱线分开的核磁共振氢谱信号,根据化学位移差异进行手性识别。
11、本发明的有益效果为:
12、以柠檬酸作为非手性碳源,(s)-(-)-α-甲基苄胺作为手性配体,采用微波辅助一步法制备了s-(-)-α-甲基苄胺手性碳量子点,并通过透射电子显微镜(tem)、傅立叶变换红外光谱(ftir)、核磁共振光谱(nmr)和x射线光电子能谱(xps)表征了形貌和化学结构。
13、本发明利用核磁共振光谱法(nmr),探究了该手性碳量子点对手性药物的手性识别性能,结果表明,其对手性氨基醇类药物普萘洛尔、阿替洛尔消旋体具有一定对映异构体识别能力,具有较好的应用前景。
技术特征:
1.s-(-)-α-甲基苄胺手性碳量子点的制备方法,其特征在于,包括以下步骤:以柠檬酸作为非手性碳源,(s)-(-)-α-甲基苄胺作为手性配体,采用微波辅助一步法制备得到所述s-(-)-α-甲基苄胺手性碳量子点。
2.如权利要求1所述的s-(-)-α-甲基苄胺手性碳量子点的制备方法,其特征在于,包括以下步骤:
3.如权利要求2所述的s-(-)-α-甲基苄胺手性碳量子点的制备方法,其特征在于,包括以下步骤:
4.权利要求1-3任一项制备方法得到的s-(-)-α-甲基苄胺手性碳量子点。
5.权利要求4所述的s-(-)-α-甲基苄胺手性碳量子点在手性氨基醇类化合物的手性识别中的应用。
6.权利要求5所述的s-(-)-α-甲基苄胺手性碳量子点在普萘洛尔或阿替洛尔对映体的手性识别中的应用。
7.如权利要求6所述的应用,其特征在于,包括以下步骤:将普萘洛尔或阿替洛尔对映体与s-(-)-α-甲基苄胺手性碳量子点混合后测定出谱线分开的核磁共振氢谱信号,根据化学位移差异进行手性识别。
技术总结
本发明属于手性识别和检测技术领域,具体涉及S‑(‑)‑α‑甲基苄胺手性碳量子点的制备方法及应用,所述制备方法包括以下步骤:以柠檬酸作为非手性碳源,(S)‑(‑)‑α‑甲基苄胺作为手性配体,采用微波辅助一步法制备得到所述S‑(‑)‑α‑甲基苄胺手性碳量子点。本发明利用核磁共振光谱法(NMR),探究了所述手性碳量子点对手性药物的手性识别性能,结果表明,其对手性氨基醇类药物普萘洛尔、阿替洛尔消旋体具有一定对映异构体识别能力,具有较好的应用前景。
技术研发人员:杨克迪,陈嘉恩,杨家興,赵振博,卢强,葛利
受保护的技术使用者:广西大学
技术研发日:
技术公布日:2024/2/29
- 还没有人留言评论。精彩留言会获得点赞!