包封的制作方法
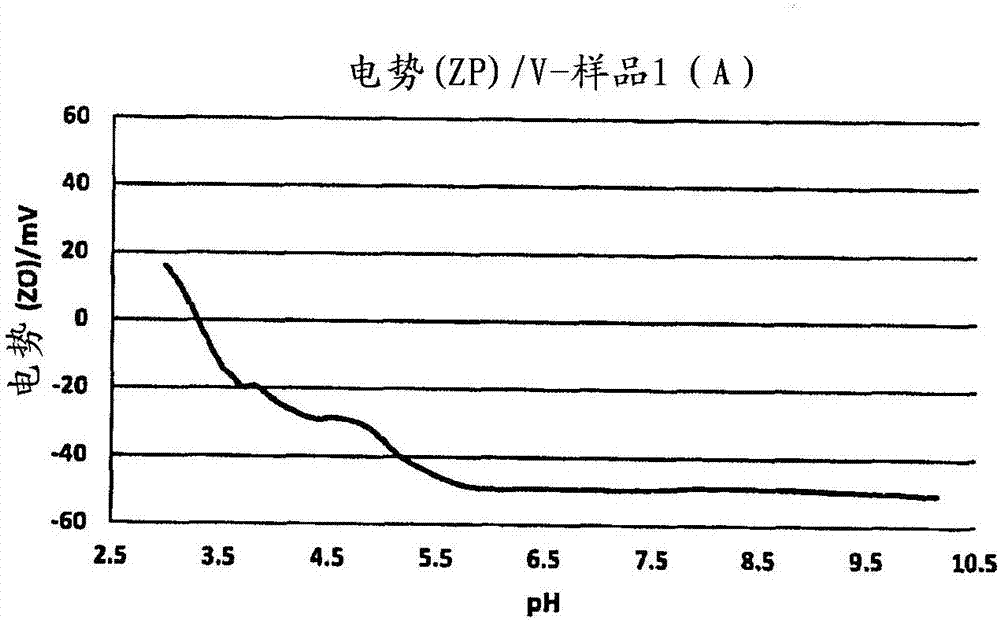
发明领域本发明涉及胶囊制备方法以及通过这样的方法制备的微胶囊。相关技术的说明用于微囊化的各种方法以及示例性的方法和物质列在schwantes(美国专利号6,592,990)、nagai等人(美国专利号4,708,924)、baker等人(美国专利号4,166,152)、wojciak(美国专利号4,093,556)、matsukawa等人(美国专利号3,965,033)、matsukawa(美国专利号3,660,304)、ozono(美国专利号4,588,639)、irgarashi等人(美国专利号4,610,927)、brown等人(美国专利号4,552,811)、scher(美国专利号4,285,720)、shioi等人(美国专利号4,601,863)、kiritani等人(美国专利号3,886,085)、jahns等人(美国专利号5,596,051和5,292,835)、matson(美国专利号3,516,941),chao(美国专利号6,375,872)、foris等人(美国专利号4,001,140;4,087,376;4,089,802和4,100,103)、greene等人(美国专利号2,800,458;2,800,457和2,730,456)、clark(美国专利号6,531,156)、saeki等人(美国专利号4,251,386和4,356,109)、hoshi等人(美国专利号4,221,710)、hayford(美国专利号4,444,699)、hasler等人(美国专利号5,105,823)、stevens(美国专利号4,197,346)、riecke(美国专利号4,622,267)、greiner等人(美国专利号4,547,429)和tice等人(美国专利号5,407,609)等中且如herbig在kirk-othmerencyclopediaofchemicaltechnology第16卷第438-463页中名称为“microencapsulation”的章节中教导的。用于微胶囊制备的其它有用的方法为∶foris等人,美国专利号4,001,140和4,089,802描述了脲和甲醛之间的反应;foris等人,美国专利号4,100,103描述了三聚氰胺和甲醛之间的反应;和英国专利号2,062,570描述了一种微胶囊的制备方法,所述微胶囊具有通过在苯乙烯磺酸的存在下聚合三聚氰胺和甲醛制备的壁。由脲醛树脂和/或三聚氰胺甲醛树脂形成微胶囊公开在美国专利号foris等人,4,001,140;foris等人,4,089,802;foris等人,4,100,103;foris等人,4,105,823;和hayford,4,444,699中。烷基丙烯酸酯-丙烯酸共聚物在brown等人的美国专利号4,552,811中有教导。在整个本申请中描述的每篇专利都通过援引其各自提供关于微囊化方法和物质的指导的程度并入本文。界面聚合是一种方法,其中在两相之间的界面形成微胶囊壁或聚酰胺、环氧树脂、聚氨酯、聚脲等。riecke的美国专利号4,622,267公开了一种用于制备微胶囊的界面聚合技术。首先,将核芯物质溶于溶剂中,并加入溶于溶剂混合物中的脂肪族二异氰酸酯。接着,加入对于脂肪族二异氰酸酯而言的非溶剂直到刚好勉强达到浊度点。然后,将有机相在水溶液中乳化,并将活性胺加入到水相中。所述胺扩散到界面,其中其与二异氰酸酯反应,形成聚合的聚氨酯壳。一种用于将微溶于水的盐包封在聚氨酯壳中的类似技术公开在greiner等人的美国专利号4,547,429中。matson的美国专利号3,516,941教导了聚合反应,其中将要包封的物质或核芯物质溶于分散在水相中的有机、疏水性油相中。所述水相具有形成氨基塑料(胺和醛)树脂的已溶物质,当聚合时其形成微胶囊的壁。使用高剪切搅拌制备微小油滴的分散液。加入酸催化剂引发在水相内形成氨基塑料树脂的缩聚,导致形成不溶于两相中的氨基塑料聚合物。随着聚合的进展,氨基塑料聚合物与水相分离,并沉积在油相的分散液滴表面上,在两相的界面处形成胶囊壁,从而包封核芯物质。脲醛(uf)、脲-间二苯酚-甲醛(urf)、脲-三聚氰胺-甲醛(umf)和三聚氰胺-甲醛(mf),胶囊形成以类似的方式进行。在界面聚合中,形成胶囊壁的物质在不同的相中,一种在水相中且另一种在油相中。聚合在相边界发生。因此,聚合胶囊壳壁在两相的界面形成,从而包封核芯物质。聚酯、聚酰胺和聚脲胶囊的壁形成通常也经由界面聚合进行。jahns的美国专利号5,292,835教导了聚合丙烯酸或甲基丙烯酸的酯与多官能单体。特别地示例的是聚乙烯吡咯烷酮与丙烯酸酯比如二丙烯酸丁二醇酯或甲基丙烯酸甲酯和自由基引发剂一起的反应。一般微囊化过程可以被视为是一系列步骤。首先,将要包封的核芯物质通常乳化或分散在合适的分散介质中。该介质通常是水性,但是参与形成富含聚合物的相。最经常地,所述介质是预期胶囊壁物质的溶液。改变该介质的溶剂特征,以便引起壁物质的相分离。壁物质由此包含在液相中,其也被分散在与预期胶囊核芯物质相同介质中。液体壁物质相本身沉积为分散的内相液滴或胶囊核芯物质周围的连续涂层。然后,将该壁物质固化。该过程一般被称为凝聚。根据本发明的胶囊用于多种胶囊内含物(“核芯物质”),包括作为举例而不限于内相油、溶剂油、相变化物质、润滑剂、染料、香料、芳香剂、清洁油、抛光油、芳香剂、营养物、甜味剂、发色团、药物、肥料、除草剂、生物学活性物、香味剂(scents)等。微胶囊核芯物质可以包括其改变流变学或流动特性或延长架存期或产品稳定性的物质。作为核芯物质的精油可以包括例如作为举例的冬绿油、肉桂油、丁香油、柠檬油、白柠檬油、橙油、薄荷油等。染料可以包括荧烷类(fluorans)、内酯类(lactones)、吲哚红、i6b、隐色染料(leucodye),全都是作为举例而非限制。核芯物质通常应当可分散或足够可溶于胶囊内相物质(即内相油)中或可溶于或可分散在内相油中溶解或分散的单体或低聚物中。核芯物质优选地为液体,但是可以是固体,取决于所选择的物质,并且适当地调节温度以引起分散。jabs等人的美国专利号4,947,152教导具有聚脲壁的微胶囊。所述壁为芳香族异氰酸酯与异氰酸酯反应基团的反应产物。异氰酸酯反应基团可以包括二胺或多胺,比如n-羟基乙基乙烯基二胺、乙烯基-1,2-二胺。hotz等人的美国专利公布2013/0089590教导了具有聚脲壁的芳香微胶囊。壳为至少两个双官能异氰酸酯和双官能胺的反应产物。ep1693104maruyyama公开了具有从多官能异氰酸酯与多官能胺缩聚获得的聚氨酯或聚脲壁的微胶囊。schwantes的美国专利公布2009/0274905教导阳离子微胶囊颗粒,其中壁为胺丙烯酸酯与多官能甲基丙烯酸酯在酸和引发剂存在下的反应产物;或者酸丙烯酸酯和多官能(甲基)丙烯酸酯在碱和引发剂存在下的反应产物。现有技术需要稳固(robust)、随时间推移保持胶囊内含物或直到破裂或以其它方式使可渗透才释放内含物的聚脲或氨基甲酸乙酯类型微胶囊。上述参考文献没有教导可以获得如下改善的微胶囊,其包含核芯,壳为包含异氰酸酯的第一组分和包含水分散性低聚多官能胺(甲基)丙烯酸酯与羧基烷基(甲基)丙烯酸酯一起的第二组分的反应混合物的产物,以得到抗破坏和抗溶剂的稳固的微胶囊。所述微胶囊用于多种挑战性环境,比如用于织物增强剂、洗衣、相变化及其它工业和商业应用。定义如本文使用的,提及术语“(甲基)丙烯酸酯”或“(甲基)丙烯酸”应当理解为指所指定单体、低聚物和/或预聚物的丙烯酸酯和甲基丙烯酸酯形式,(例如"(甲基)丙烯酸烯丙酯"指甲基丙烯酸烯丙酯和丙烯酸烯丙酯都是可行的,类似地提及(甲基)丙烯酸的烷基酯指丙烯酸的烷基酯和甲基丙烯酸的烷基酯都是可行的,类似地聚(甲基)丙烯酸酯指聚丙烯酸酯和聚甲基丙烯酸酯都是可行的)。除非另有说明,否则本文的每个烷基部分都可以来自c1至c8,或者甚至c1至c24。聚(甲基)丙烯酸酯物质旨在涵盖广泛的聚合物物质,包括例如聚酯聚(甲基)丙烯酸酯、氨基甲酸乙酯和聚氨酯聚(甲基)丙烯酸酯(特别是由(甲基)丙烯酸羟基烷基酯与聚异氰酸酯或氨基甲酸乙酯聚异氰酸酯反应制备的那些)、氰基丙烯酸甲酯、氰基丙烯酸乙酯、二(甲基)丙烯酸二甘醇酯、三羟甲基丙烷三(甲基)丙烯酸酯、二(甲基)丙烯酸乙二醇酯、(甲基)丙烯酸烯丙酯、(甲基)丙烯酸缩水甘油酯、(甲基)丙烯酸酯官能化硅氧烷、二(甲基)丙烯酸二甘醇酯、二(甲基)丙烯酸三甘醇酯、和二(甲基)丙烯酸四甘醇酯、二(甲基)丙烯酸二丙二醇酯、二(甲基)丙烯酸聚乙二醇酯、二(甲基)丙烯酸二(戊二醇)酯、二(甲基)丙烯酸乙烯基酯、二(甲基)丙烯酸新戊二醇酯、三羟甲基丙烷三(甲基)丙烯酸酯、乙氧基化的双酚a二(甲基)丙烯酸酯、双酚a二(甲基)丙烯酸酯、二(甲基)丙烯酸二甘油酯、二氯丙烯酸四甘醇酯、二(甲基)丙烯酸1,3-丁二醇酯、二(甲基)丙烯酸新戊酯、三羟甲基丙烷三(甲基)丙烯酸酯、二(甲基)丙烯酸聚乙二醇酯和二(甲基)丙烯酸二丙二醇酯和各种多官能(甲基)丙烯酸酯,以及多官能胺(甲基)丙烯酸酯。也可以有利地使用单官能丙烯酸酯,即仅含有一个丙烯酸酯基团的那些。典型的单丙烯酸酯包括(甲基)丙烯酸2-乙基己酯、(甲基)丙烯酸2-羟基乙酯、(甲基)丙烯酸氰基乙酯、(甲基)丙烯酸2-羟基丙酯、(甲基)丙烯酸对二甲基氨基乙酯、(甲基)丙烯酸月桂酯、(甲基)丙烯酸环己酯、(甲基)丙烯酸四氢糠酯、(甲基)丙烯酸氯苄酯、(甲基)丙烯酸氨基烷基酯、各种(甲基)丙烯酸烷基酯和(甲基)丙烯酸缩水甘油酯。当然,也可使用(甲基)丙烯酸酯或它们衍生物的混合物,以及一种或多种(甲基)丙烯酸酯单体、低聚物和/或预聚物或它们的衍生物与其它可共聚单体(包括丙烯腈和甲基丙烯腈)的组合。附图简述图1给出了实施例8的微胶囊在己烷中渗漏的图表。图2、3、4、5和6给出了实施例8中描述的样品的ζ电势的图表。发明简述本发明包括包含核芯和包围核芯物质的壳的微胶囊,所述壳包括包含异氰酸酯的第一组分和包含一种或多种聚(甲基)丙烯酸酯,更特别是多官能胺(甲基)丙烯酸酯的第二组分的反应产物,其中所述多官能胺(甲基)丙烯酸酯选择为具有极性的且与异氰酸酯反应。任选地,但优选地,将羧基烷基(甲基)丙烯酸酯与多官能胺(甲基)丙烯酸酯混合。在本发明中,胶囊壁物质具有作为主要组分的聚氨酯或聚脲和作为次要组分的丙烯酸酯预聚物或聚合物。在一个方面,本发明包括包含油溶性或分散性有益剂核芯物质和包围所述有益剂核芯物质的壳,所述壳包含由第一组分异氰酸酯和第二组分胺形成的聚脲,所述胺包括聚(甲基)丙烯酸酯,更特别地,(甲基)丙烯酸烷基氨基烷基酯和多官能(甲基)丙烯酸酯与羧基烷基(甲基)丙烯酸酯反应的反应产物。在另一个方面,本发明包括微胶囊,其中壳包含异氰酸酯和多官能胺(甲基)丙烯酸酯的反应产物。在进一步的实施方案中,微胶囊的第二组分多官能胺(甲基)丙烯酸酯为低聚物,或者可选地,异氰酸酯为低聚物。第一组分的异氰酸酯基与第二组分的胺或羟基的摩尔比范围为0.5:1至约20:1。核芯包括有益剂核芯物质。在仍然进一步的实施方案中,本发明包括一种制备有益剂递送颗粒的方法,所述方法包括在一个或多个步骤中加热乳剂,所述乳液是通过乳化第一组合物的组合制备的,所述第一组合物是通过混合水相1、水相2和水相3形成的;所述水相1包含水和引发剂;所述水相2包含水、(甲基)丙烯酸羟基烷基酯和多官能(甲基)丙烯酸酯;所述水相3包含水和羧基烷基(甲基)丙烯酸酯和碱;和第二组合物,所述第二组合物包括包含异氰酸酯和核芯物质的油相。在进一步的实施方案中,本发明包括微胶囊,其包含油溶性或分散性有益剂核芯物质和包围所述有益剂核芯物质的壳。所述壳包括由第一组分异氰酸酯和第二组分多元醇形成的聚氨酯。多元醇包括(甲基)丙烯酸羟基酯和多官能(甲基)丙烯酸酯与羧基烷基(甲基)丙烯酸酯或季铵丙烯酸盐一起的反应产物。在一个实施方案中,多元醇为(甲基)丙烯酸羟基酯,更特别地为(甲基)丙烯酸羟基烷基酯比如(甲基)丙烯酸羟基乙酯。可选地,壳包括异氰酸酯和多官能(甲基)丙烯酸羟基酯的反应产物。在一个实施方案中,异氰酸酯可以选自异氟尔酮二异氰酸酯、4,4'-亚甲基二苯基二异氰酸酯、2,2'-亚甲基二苯基二异氰酸酯和2,4'-亚甲基二苯基二异氰酸酯。可选地,多元醇可以是其中每个烷基部分独立地为c1至c8或甚至c1至c24的(甲基)丙烯酸羟基烷基酯。在进一步的实施方案中,(甲基)丙烯酸羟基酯选自(甲基)丙烯酸羟基烷基酯、(甲基)丙烯酸烷二醇酯和1,3-二甘油酯二丙烯酸甘油酯。本发明的微胶囊在环境比如与溶剂、洗涤剂、洗发剂、织物柔软剂和表面清洁剂接触的环境中显示高强度和低渗漏。因此,本发明的微胶囊适用于这样的产品中,其能够在这样的环境中保持完整(survive)。另外,比如当核芯选择为相变材料(潜热材料)时,本发明的微胶囊可以有利地应用于产品比如床垫、枕头、床、织物、运动器材、医疗装置、建筑产品、结构产品、供热和通风应用(hvac)、再生能源应用、太阳能板、衣服、运动器材表面(athleticsurfaces)、汽车、航空、鞋、美容护理、洗衣和太阳能产品中。本发明公开了具有带表面电荷的壁的微胶囊,所述微胶囊是通过包括下述步骤的方法制备的∶将引发剂、具有一个或多个-oh、-nh2或-nh-基团的交联官能单体和具有阴离子或阳离子基团的电荷官能单体分散在一个或多个水相中,所述阴离子或阳离子基团可选自羧基、磺酸基、季铵基团或其它带电荷基团。所述单体在一个或多个水相中预反应,并与水分散性多官能(甲基)丙烯酸酯单体混合。乳液是通过使用高剪切搅拌将包含异氰酸酯和有益剂核芯物质的油相乳化到水相或多个水相中形成的。任选地,可以加入另外的交联剂,比如含有2个或更多个的伯胺或仲胺基团的化合物。通过加热或光化辐射一定时间,且温度或辐射足以形成包围有益剂核芯物质的微胶囊壁,使预反应单体、水分散性多官能(甲基)丙烯酸酯单体和油相的混合乳液进一步反应。有利地,具有-oh、-nh2或-nh-基团的交联官能单体可以为胺,比如(甲基)丙烯酸烷基氨基烷基酯。可选地,具有-oh、-nh2或-nh-基团的交联官能单体可以为羟基,比如存在于(甲基)丙烯酸羟基酯中的羟基。任选地,在微胶囊壁形成之后,可以将形成的微胶囊与水相或连续相分离,比如通过倾析、脱水、离心、喷雾干燥、蒸发、冷冻干燥或其它溶剂去除或干燥方法。详细说明本发明公开了形成微胶囊群的组合物和方法。所述微胶囊包括油溶性或分散性有益剂核芯物质和包围所述有益剂核芯物质的壳。所述壳包括异氰酸酯和多官能胺(甲基)丙烯酸酯的反应产物。多官能胺(甲基)丙烯酸酯可以选择为极性的且与异氰酸酯反应的。本发明的方法是基于形成水包油型乳液以产生包封。本发明包括微胶囊,它包含油溶性或分散性有益剂核芯物质和包围所述有益剂核芯物质的壳,所述壳包括由第一组分异氰酸酯和第二组分形成的聚脲,所述第二组分为具有交联官能团比如-oh、-nh2或-nh-的交联官能聚合物或低聚物。当第二组分为胺时,所述胺包括(甲基)丙烯酸烷基氨基烷基酯和多官能(甲基)丙烯酸酯与带电荷官能单体比如(甲基)丙烯酸羧基烷基酯或季铵丙烯酸盐的反应产物。选择水溶性且具有交联官能团的交联官能单体,比如例如甲基丙烯酸羟乙酯、甲基丙烯酸2-叔-(丁基氨基)乙酯或甲基丙烯酸2-氨基乙酯。也可以选择水溶性的带电荷官能单体,比如2-(甲基丙烯酰基氧基乙基)三甲基氯化铵或(甲基)丙烯酸羧基烷基酯。还选择水分散性的多官能单体,并且选自物质比如乙氧基化的三羟甲基丙烷三丙烯酸酯或二丙烯酸聚乙二醇酯或二甲基丙烯酸聚乙二醇酯。为了产生嵌段聚合,将采用引发剂和反应性交联官能单体和多官能丙烯酸酯的预引发(pre-initiation)步骤应用于预加热步骤中,形成丙烯酸酯预聚物。该预聚物与带电荷官能单体进一步反应,得到嵌段聚合物。进行乳化,而不必实质性加入乳化剂比如聚乙烯醇。因此,乳化剂是任选的。任选地,可以在乳化之后加入另外的交联剂。这样的化合物包含两个或更多个伯胺或仲胺基团,并且可以选自本领域已知的各种胺交联剂,包括而不限于交联剂比如乙二胺、二乙烯三胺、三乙烯四胺、四乙烯五胺或五乙烯六胺。其它示例性的交联剂可以包括n-(甲基异戊基)乙二胺、n-(苄基)乙二胺、n-(2-乙基己基)乙二胺、n-(异丙基)乙二胺、n-(4-甲基苄基)乙二胺、n-(3-甲基苄基)乙二胺、n-(2-甲基苄基)乙二胺、n-(4-甲氧基苄基)乙二胺、n-(3-甲氧基苄基)乙二胺、n-(2-甲氧基苄基)乙二胺、n-(2-甲基丙基)乙二胺、n-(2-甲基丁基)乙二胺、n-(甲基-丙基)乙二胺、n-(仲-丁基)乙二胺、n-(仲-苯基乙基)乙二胺、n-(叔-丁基)乙二胺、n,n”'-双-(甲基异戊基)三乙烯四胺、n,n”'-双-(苄基)三乙烯四胺、n,n”'-双-(2-乙基己基)三乙烯四胺、n,n”'-双-(异丙基)三乙烯四胺、n,n”'-双-(4-甲基苄基)三乙烯四胺、n,n”'-双-(3-甲基苄基)三乙烯四胺、n,n”'-双-(2-甲基苄基)三乙烯四胺、n,n”'-双-(4-甲氧基苄基)三乙烯四胺、n,n”'-双-(3-甲氧基苄基)三乙烯四胺、n,n”'-双(2-甲氧基苄基)三乙烯四胺、n,n”'-双-(2-甲基丙基)三乙烯四胺、n,n”'-双-(2-甲基丁基)三乙烯四胺、n,n”'-双-(甲基-丙基)三乙烯四胺、n,n”'-双-(仲-丁基)三乙烯四胺、n,n”'-双-(仲-苯基乙基)三乙烯四胺、n,n”'-双-(叔-丁基)三乙烯四胺、n,n'-双-(甲基异戊基)乙二胺、n,n'-双-(苄基)乙二胺、n,n'-双-(2-乙基己基)乙二胺、n,n'-双-(4-甲基苄基)乙二胺、n,n'-双-(异丙基)乙二胺、n,n'-双-(3-甲基苄基)乙二胺。交联剂可以单独或者作为交联剂的混合物使用。另外的交联剂是本领域已知,比如在专利公布us20080090922中教导的,将其并入本文作为参考。在另一个方面,本发明包括微胶囊,其中壳包含异氰酸酯和多官能胺(甲基)丙烯酸酯的反应产物。有用的有益剂核芯物质包括香料原料比如醇、酮、醛、酯、醚、腈、链烯、芳香剂、芳香剂增溶剂、精油、相变材料、润滑剂、着色剂、冷却剂、防腐剂、抗微生物或抗真菌活性剂、除草剂、抗病毒活性剂、杀菌活性剂、抗氧剂、生物活性剂、除臭剂、软化剂、湿润剂、剥离剂、紫外线吸收剂、自愈组合物、腐蚀抑制剂、防晒剂、硅氧烷油、蜡、烃、高级脂肪酸、精油、脂质、皮肤冷却剂、维生素、防晒剂、抗氧剂、甘油、催化剂、漂白颗粒、二氧化硅颗粒、恶臭减少剂(malodorreducingagents)、染料、增亮剂、抗菌活性剂、止汗活性剂、阳离子聚合物及其混合物。用作核芯物质的相变材料可以包括作为举例而不限于具有13至28个碳原子的石蜡烃、各种烃类如正二十八烷、正二十烷、正二十六烷、正二十五烷、正二十四烷、正二十三烷、正二十二烷、正二十一烷、正二十烷、正十九烷、十八烷、正十七烷、正十六烷、正十五烷、正十四烷、正十三烷。相材料可以可选任选地另外包括晶体材料,比如2,2-二甲基-1,3-丙二醇、2-羟甲基-2-甲基-1,3-丙二醇、直链或支链烃的酸比如花生酸和酯比如棕榈酸甲酯、脂肪醇、及其混合物。交联官能单体可以选自甲基丙烯酸叔丁基氨基乙酯、甲基丙烯酸叔丁基氨基丙酯、甲基丙烯酸正丁基氨基乙酯、甲基丙烯酸二乙基氨基乙酯、甲基丙烯酸二甲基氨基乙酯、甲基丙烯酸二异丙基氨基乙酯、甲基丙烯酸二丁基氨基乙酯、甲基丙烯酸二丙基氨基乙酯、甲基丙烯酸叔戊基氨基乙酯、甲基丙烯酸叔己基氨基乙酯、甲基丙烯酸叔丁基氨基丙酯、甲基丙烯酸二乙基氨基丙酯和甲基丙烯酸二甲基氨基丙酯。多官能丙烯酸酯或甲基丙烯酸酯单体或低聚物可以包括一-;二-;三-;四-;五-;六-;七-;或八-官能丙烯酸酯、甲基丙烯酸酯和多官能聚氨酯丙烯酸酯和环氧丙烯酸酯。单体应当理解为包括其低聚物。任选地,可以将抑制剂比如对苯二酚加入到胶囊中的单体和引发剂混合物中以防止过早聚合。本发明中有用的是二-和多-官能(甲基)丙烯酸酯、双官能(甲基)丙烯酸酯、多官能(甲基)丙烯酸酯、双官能氨基甲酸乙酯丙烯酸酯、多官能氨基甲酸乙酯丙烯酸酯以及多官能和双官能环氧丙烯酸酯单体和低聚物,单独使用或作为掺和物组合使用。在可选的实施方案中,任选地,将二-和多-官能丙烯酸酯、甲基丙烯酸酯、氨基甲酸乙酯丙烯酸酯和环氧胺丙烯酸酯与单官能丙烯酸酯、甲基丙烯酸酯、氨基甲酸乙酯丙烯酸酯和环氧丙烯酸酯进一步混合。用于本发明的合适的异氰酸酯可以选自单体和低聚物和掺合物,并且可以为c2-c24直链、支链、环状、芳香族的或其摻合物。适合使用的异氰酸酯包括但不限于二异氰酸酯,比如异氟尔酮二异氰酸酯,也称为3,3,5-三甲基-5-异氰酸基-甲基-环己基异氰酸酯或ipdi;氢化的物质,比如亚环己基二异氰酸酯、4,4'-亚甲基二环己基二异氰酸酯、4,4'-亚甲基二苯基二异氰酸酯(“mdi”),2,2'-亚甲基二苯基二异氰酸酯、2,4'-亚甲基二苯基二异氰酸酯(mdi)、芳烷基二异氰酸酯比如四甲基二甲苯基二异氰酸酯、聚亚甲基异氰酸酯比如1,4-四亚甲基二异氰酸酯、1,5-五亚甲基二异氰酸酯、1,6-六亚甲基二异氰酸酯(hmdi)、1,7-七亚甲基二异氰酸酯、2,2,4-三甲基六亚甲基二异氰酸酯和2,4,4-三甲基六亚甲基二异氰酸酯、1,10-十亚甲基二异氰酸酯和2-甲基-1,5-五亚甲基二异氰酸酯;及其混合物。异氰酸酯可以包括芳香族异氰酸酯,不限于亚苯基二异氰酸酯、甲苯二异氰酸酯、二甲苯二异氰酸酯、1,5-萘二异氰酸酯、氯亚苯基2,4-二异氰酸酯、二甲苯二异氰酸酯、联甲氧基苯胺二异氰酸酯、联甲苯胺二异氰酸酯、烷基化苯二异氰酸酯、亚甲基-插入的(interrupted)芳香族二异氰酸酯比如亚甲基二苯基二异氰酸酯、4,4'-异构体(mdi)包括烷基化类似物比如3,3'-二甲基-4,4'-二苯甲烷二异氰酸酯、聚合亚甲基二苯基二异氰酸酯及其混合物。本发明同样应用于类似的聚氨酯组合物和方法。可以形成包含油溶性或分散性有益核芯物质的微胶囊。包围具有有益剂物质的油核芯的微胶囊壳则是由第一组分异氰酸酯和第二组分多元醇形成的聚氨酯。该组合中的多元醇为具有交联官能团比如-oh的交联官能单体,比如(甲基)丙烯酸羟基酯和多官能(甲基)丙烯酸酯与带电荷官能单体比如(甲基)丙烯酸羧基酯或季铵丙烯酸盐一起的反应产物。(甲基)丙烯酸羧基酯和/或季铵丙烯酸盐提供给得到的聚氨酯嵌段共聚物带电荷区域或带电荷侧基,有助于使所述聚合物到达相界面,引起包围有益剂的微胶囊壳形成,所述有益剂溶解或分散在油相液滴中。通过经由所得聚合物的带电荷区域或带电荷侧基的化学连接,本发明使定制表面电荷成为可能。表面电荷可以改善微胶囊在基质比如织物、皮肤、毛发、纤维或其它表面上的沉积。表面电荷也可以有利地应用于改善微胶囊在表面比如泡沫或垫层料上的粘附。当期望高固体、团块或干燥粉末的微胶囊时,表面电荷也可以有利地适合产生凝聚物,以促进过滤容易性。如果期望,可以从水性介质分离微胶囊。浆料可以作为脱水团块使用,或者以干粉形式使用,取决于应用。多元醇可以为(甲基)丙烯酸羟基酯,选自(甲基)丙烯酸羟基烷基酯,比如(甲基)丙烯酸羟基乙酯或(甲基)丙烯酸羟基丙酯。烷基可以为c1-c8碳中任一个。(甲基)丙烯酸羟基酯也可以为羟基取代的(甲基)丙烯酸酯比如(甲基)丙烯酸烷二醇酯,和羟基-取代的二-和三-丙烯酸酯1,3-二甘油酯二丙烯酸甘油酯。丙烯酸酯引发剂被能量活化指当进行加热或其它能量输入比如光化辐射或离子束时产生自由基。优选的引发剂包括过氧引发剂、偶氮引发剂、过氧化物和化合物比如2,2'-偶氮二甲基丁腈、二苯甲酰基过氧化物。更特别地,且不限于可以选自下述引发剂的自由基引发剂:偶氮或过氧引发剂,比如过氧化物、二烷基过氧化物、烷基过氧化物、过氧化酯、过氧化碳酸酯、过氧化酮和过氧化二碳酸酯、2,2'-偶氮双(异丁基腈)、2,2'-偶氮双(2,4-二甲基戊腈(pentanenitrile))、2,2'-偶氮双(2,4-二甲基戊腈(valeronitrile))、2,2'-偶氮双(2-甲基丙腈)、2,2'-偶氮双(甲基丁腈)、1,1'-偶氮双(环己烷甲腈)、1,1'-偶氮双(氰基环己烷)、苯甲酰基过氧化物、癸酰基过氧化物;月桂酰基过氧化物;苯甲酰基过氧化物、二(正-丙基)过氧化二碳酸酯、二(仲-丁基)过氧化二碳酸酯、二(2-乙基己基)过氧化二碳酸酯、1,1-二甲基-3-羟丁基过氧化新癸酸酯、α-枯基过氧化新庚酸酯、叔戊基过氧化新癸酸酯、叔丁基过氧化新癸酸酯、叔戊基过氧化特戊酸酯、叔丁基过氧化特戊酸酯、2,5-二甲基-2,5-二(2-乙基己酰基过氧基)己烷、叔戊基过氧基-2-乙基己酸酯、叔丁基过氧基-2-乙基己酸酯、叔丁基过氧化乙酸酯、二戊基过氧化乙酸酯、叔丁基过氧化物、二叔戊基过氧化物、2,5-二甲基-2,5-二(叔丁基过氧基)己炔-3、异丙基苯过氧化氢、1,1-二(叔丁基过氧基)-3,3,5-三甲基环己烷、1,1-二(叔丁基过氧基)-环己烷、1,1-二(叔戊基过氧基)-环己烷、乙基-3,3-二(叔丁基过氧基)-丁酸酯、叔戊基过苯甲酸酯、叔丁基过苯甲酸酯、乙基3,3-二(叔戊基过氧基)-丁酸酯等。也可以使用引发剂的混合物。引发剂是商业可获得的,比如vazo引发剂,其通常标明引发剂的分解温度。优选地,选择具有约50℃或更高的分解点的引发剂。使用有用的多种引发剂,呈在油相中的混合物,或在油相或水相的任一个中的混合物。优选地,选择在各个步骤:胶囊壁物质的预聚、壁形成和硬化或聚合中错开分解温度的引发剂。例如,油相中的第一引发剂可以在55℃下分解以促进预聚物形成,第二引发剂可以在60℃下分解以促进形成壁物质。任选地,第三引发剂可以在65℃下分解以促进胶囊壁物质的聚合。引发剂的总量通常可以低至0.1重量%或高达10重量%。用于本文的目的,术语分散相或油相可互换地使用,并且可以选自烃类,更特别是烃溶剂类,并且所述溶剂可以包括作为举例而不限于乙基二苯基甲烷、丁基二苯基乙烷、苄基二甲苯、烷基联苯比如丙基联苯和丁基联苯、邻苯二甲酸二烷基酯例如邻苯二甲酸二丁酯、邻苯二甲酸二辛酯、邻苯二甲酸二壬酯和邻苯二甲酸双十三烷基酯;2,2,4-三甲基-1,3-戊二醇-二异丁酯、烷基苯比如十二烷基苯;以及羧酸酯类,醚类或酮类,比如二芳基醚类、二(芳烷基)醚和芳基芳烷基醚,醚类比如二苯醚、二苄醚和苯基苄基醚、液体高级烷基酮类(具有至少9个碳原子)、烷基或芳烷基苯甲酸酯例如苯甲酸苄酯、烷基化的萘类比如二丙基萘、部分氢化的三联苯类;高沸点的直链或支链烃类、芳烃类和烷芳基烃类,比如甲苯,植物油比如低芥酸菜子油、大豆油、玉米油、向日葵油或棉籽油,衍生自低芥酸菜子油、大豆油、棉籽油、玉米油、向日葵油、松油、柠檬油、橄榄油的酯交换的脂肪酸的甲基酯,或油酸的甲酯、植物油、植物油的酯,例如大豆甲酯,10至13个碳的直链饱和的链烷脂肪族烃;c8-c42酯、己酸乙酯、庚酸甲酯、丁酸丁酯、苯甲酸甲酯、甲基比如壬酸酯(nonoate)、癸酸甲酯、十二烷酸甲酯、辛酸甲酯、月桂酸甲酯、肉豆蔻酸甲酯、棕榈酸甲酯、硬脂酸甲酯、庚酸乙酯、辛酸乙酯、壬酸乙酯、癸酸乙酯、十二烷酸乙酯、月桂酸乙酯、豆蔻酸乙酯、棕榈酸乙酯、硬脂酸乙酯、肉豆蔻酸异丙酯、棕榈酸异丙酯、棕榈酸乙基己酯、月桂酸异戊酯、月桂酸丁酯、辛酸辛酯、癸酸癸酯、硬脂酸丁酯、月桂酸月桂基酯、棕榈酸硬脂基酯、硬脂酸硬脂基酯、山嵛酸硬脂基酯和山嵛酸山嵛酯。也可以使用上述的混合物。一般的稀释剂比如直链烃也可以与溶剂、或溶剂的掺合物混合。溶剂的选择是基于疏水性和分散或溶剂化异氰酸酯的能力。本发明的方法提供了稳固的有益剂递送颗粒。所述有益剂递送颗粒为包围核芯物质的壳的微胶囊。形成微胶囊的方法包括形成不同的水相,优选地包括提供水相1、水相2和水相3的第一组合物。水相1包括水和引发剂。水相2包括水、水溶性或分散性的胺(甲基)丙烯酸酯和多官能(甲基)丙烯酸酯。水相3包括水和羧基取代的烷基(甲基)丙烯酸酯及任选的碱。在一个实施方案中,在第一步中,使混合的(甲基)丙烯酸酯单体预反应,形成多官能的胺(甲基)丙烯酸酯预聚物。乳液是通过在高剪切搅拌下将第二组合物乳化到第一组合物中形成的;所述第二组合物包括包含异氰酸酯和有益剂核芯物质的油相。在一个或多个步骤中,加热所述乳液形成包含异氰酸酯和多官能的胺(甲基)丙烯酸酯的反应产物的壁物质,所述壁包围有益剂核芯物质。在本发明的方法和组合物中,电荷可以被定制为在ph7下的ζ电势,ζ电势范围为+70至-70,并且在许多应用中,有利地范围为+40至65是有用的。优选的ζ电势大于+70,或大于+40,或大于-70,或者甚至大于-40。有用的ζ电势为+70至+20,或-20至-70;或者甚至ζ电势为+70至+40,或者-40至-70;或者甚至+70至+50,或者甚至-50至-70。在上下文中,“大于”或“高于”指更高的电荷值,无论正电荷还是负电荷。更多正电荷(更大正值)或更多负电荷值(更大负值)是优选的。任选地,可以包括沉积助剂,以增强微胶囊沉积或粘附不同表面,比如各种基质,包括但不限于纸、护肤织物(fabricskin)、毛发、毛巾或其它表面。沉积助剂可以包括聚(丙烯酰胺-共-二烯丙基二甲基氯化铵)、聚(二烯丙基二甲基氯化铵)、聚乙烯亚胺、阳离子多胺、聚[(3-甲基-1-乙烯基咪唑氯化鎓)-共-(1-乙烯基吡硌烷酮)]、丙烯酸和二烯丙基二甲基氯化铵的共聚物、阳离子瓜尔胶、瓜尔胶、比如在美国公开20150030557(并入本文作为参考)中描述的聚有机硅氧烷。在一个进一步的实施方案中,上述微胶囊可以包括沉积助剂,并且在一个进一步的方面,沉积助剂涂布微胶囊壳的外表面。在一个进一步的方面,沉积助剂可以包括选自下述的物质:聚(甲基)丙烯酸酯、聚(乙烯-马来酸酐)、聚胺、蜡、聚乙烯吡咯烷酮、聚乙烯吡咯烷酮共聚物、聚乙烯吡咯烷酮-丙烯酸乙酯、聚乙烯吡咯烷酮-丙烯酸乙烯酯、聚乙烯吡咯烷酮甲基丙烯酸酯、聚乙烯吡咯烷酮-乙酸乙烯酯、聚乙烯醇缩醛、聚乙烯醇缩丁醛、聚硅氧烷、聚(丙烯马来酸酐)、马来酸酐衍生物、马来酸酐衍生物的共聚物、聚乙烯醇、苯乙烯-丁二烯胶乳、明胶、阿拉伯树胶、羧甲基纤维素、羧甲基羟乙基纤维素、羟乙基纤维素、其它改性的纤维素、藻酸钠、壳聚糖、酪蛋白、果胶、改性淀粉、聚乙烯醇缩醛、聚乙烯醇缩丁醛、聚乙烯基甲基醚/马来酸酐、聚乙烯基吡咯烷酮及其共聚物、聚(乙烯基吡咯烷酮/甲基丙烯酰胺基丙基三甲基氯化铵)、聚乙烯吡咯烷酮/乙酸乙烯酯、聚乙烯基吡咯烷酮/甲基丙烯酸二甲基氨基乙酯、聚乙烯胺、聚乙烯基甲酰胺、聚烯丙基胺、以及聚乙烯胺、聚乙烯基甲酰胺和聚烯丙基胺的共聚物、及其混合物。在一个仍然进一步的方面,沉积助剂包括选下述的物质:聚(甲基)丙烯酸酯、聚(乙烯-马来酸酐)、聚胺、聚乙烯吡咯烷酮、聚乙烯吡咯烷酮-丙烯酸乙酯、聚乙烯吡咯烷酮-丙烯酸乙烯酯、聚乙烯吡咯烷酮甲基丙烯酸酯、聚乙烯吡咯烷酮-乙酸乙烯酯、聚乙烯醇缩醛、聚硅氧烷、聚(丙烯马来酸酐)、马来酸酐衍生物、马来酸酐衍生物的共聚物、聚乙烯醇、羧甲基纤维素、羧甲基羟乙基纤维素、羟乙基纤维素、聚乙烯基甲基醚/马来酸酐、聚乙烯吡咯烷酮/乙酸乙烯酯、聚乙烯基吡咯烷酮/甲基丙烯酸二甲基氨基乙酯、聚乙烯胺、聚乙烯基甲酰胺、聚烯丙基胺、以及聚乙烯胺、聚乙烯基甲酰胺和聚烯丙基胺的共聚物、及其混合物。在下述实施例中,缩写对应于下述物质:表1实施例1聚合物的制备方法∶对于水相1,将0.5克水溶性的引发剂(v50)加入到在40℃下夹套钢制反应器中的200g水中,同时以1000rpm混合,并且以100cc/min用氮气覆盖。在45分钟内,将溶液从40℃加热至75℃,保持在75℃45分钟,并且在75分钟内冷却至60℃。加入150g的水相2,其包括2.5g的至少一种多官能水分散性丙烯酸单体(sr415)和10克的至少一种具有交联官能团的水溶性丙烯酸单体(即tbaema),并且混合提高至1500rpm。在60℃下,将合并的水相混合60分钟,然后加入150g的水相3,其包括12.5克的至少一种具有表面电荷官能团的水溶性丙烯酸单体(cd9055),并将合并的溶液保持在60℃下另外30分钟。然后,在30分钟内将溶液加热到75℃,在7小时内从75℃加热至95℃,并保持在该温度下6小时,形成最终聚合物。实施例1a∶使用caframobdc6015搅拌器、finemechkgw-2205回火烧杯(temperingbeaker)(反应器)和coleparmer再循环加热水浴制备该批料。对于内相,将90g的油相置于烧杯中,并与7.78g的desmoduri(ipdi)和3.35g的mondurmr(mdi)混合,使用搅拌器(stirplate)混合直至均质。在反应器中,通过向144g的水中加入6g的丙烯酸酯阴离子聚合物(上述制备的),并用设定7℃的再循环水浴混合30分钟来开始制备水相。在混合水相之后,经1分钟,将内相加入到反应器中,并设定caframo的速度为2500rpm开始研磨,形成目标粒度(即10um)的稳定乳液。在研磨结束时,关掉搅拌器,并用z-棒代替研磨叶片。然后,将搅拌速度设定为200rpm,并将该批料在120分钟内加热至60℃,保持在60℃240分钟,并在60分钟内加热至85℃,保持在85℃240分钟。在加热循环之后,将温度设定为恢复室温。实施例1b∶使用caframobdc6015搅拌器、finemechkgw-2205回火烧杯(反应器)和coleparmer再循环加热水浴制备该批料。对于内相,将90g的油相置于烧杯中,并与7.78的desmoduri(ipdi)和3.35g的mondurmr(mdi)混合,使用搅拌器混合直至均质。在反应器中,通过向144g的水中加入6g的丙烯酸酯阴离子聚合物(上述制备的),并用设定7℃的再循环水浴混合30分钟来开始制备水相。在混合水相之后,经1分钟,将内相加入到反应器中,并设定caframo的速度为2500rpm开始研磨,形成目标粒度(即10um)的稳定乳液。在研磨结束时,关掉搅拌器,并用z-棒代替研磨叶片。然后,将搅拌速度设定为200rpm,并将该批料在120分钟内加热至60℃,保持在60℃下另外20分钟,并加入1.8g另外的交联剂(deta)。然后,使该批料保持在60℃另外120分钟,在60分钟内加热至85℃,并保持在85℃240分钟。在加热循环完成之后,将温度设定为恢复室温。实施例1c:使用caframobdc6015搅拌器、finemechkgw-2205回火烧杯(反应器)和coleparmer再循环加热水浴制备该批料。对于内相,将75g的油相置于烧杯中,并与25g的desmodurw(h12mdi)混合,使用搅拌器混合直至均质。在反应器中,通过向284.14g的水中加入18.11g的丙烯酸酯阴离子聚合物(上述制备的)和7.75g的evonikox50亲水性二氧化硅,并用设定35℃的再循环水浴混合15分钟来开始制备水相。在混合水相之后,经1分钟将内相加入到反应器中,并将caframo的速度设定为3000rpm开始研磨。在一小时的研磨结束时,关掉搅拌器,并用z-棒代替研磨叶片。然后,将速度设定为350rpm,将该批料加热至92℃,并保持在该温度12小时,之后将温度设定为恢复室温。实施例1d:对于该实验室批量生产,使用caframobdc6015搅拌器、finemechkgw-2205回火烧杯(反应器)和coleparmer再循环加热水浴制备该批料。对于内相,将240g的油置于烧杯中,并与3.0gdesmodurn3300a和7.0g的desmodurn3400混合,使用搅拌器混合直至均质。在反应器中,通过向179.0g的水中加入33.1g的丙烯酸酯阴离子聚合物(上述制备的),并用设定35℃的再循环水浴混合15分钟来开始制备水相。在混合水相之后,将caframo搅拌器升高至2000rpm,并经2分钟将内相加入到反应器中。当已经加入所有的内相之后,将caframo的速度设定为3000rpm开始研磨。在研磨结束时,关掉搅拌器,并用z-棒代替研磨叶片。然后,将速度设定为500rpm,并将再循环水浴设定为40℃两小时。然后,使水浴温度上升至60℃,并保持3小时,之后将温度设定为恢复室温。所有样品的最后包封都可以包含具有表面电荷官能团的聚丙烯酸酯/聚脲双组分壁系统。该胶囊是阴离子的,并且显示出低渗漏。实施例2聚合物的制备方法∶对于水相1,将0.825克水溶性的引发剂(v50)加入到在40℃下夹套钢制反应器中的200g水中,同时以1000rpm混合,并且以100cc/min用氮气覆盖。在45分钟内,将溶液从40℃加热至75℃,保持在75℃45分钟,并且在75分钟内冷却至60℃。加入150g的水相2,其包括2.5g的至少一种多官能水分散性丙烯酸单体(sr415)和10克的至少一种具有交联官能团的水溶性丙烯酸单体(即,羟乙基甲基丙烯酸酯(hema)),并且混合提高至1500rpm。在60℃下,将合并的水相混合60分钟,然后加入150g的水相3,其包括15克的至少一种具有表面电荷官能团的水溶性丙烯酸单体(cd9055),并将合并的溶液保持在60℃下另外30分钟。然后,在30分钟内将溶液加热到75℃,在7小时内从75℃加热至95℃,并保持在95℃下6小时,形成最终聚合物。实施例2a:使用caframobdc6015搅拌器、finemechkgw-2205回火烧杯(反应器)和coleparmer再循环加热水浴制备该批料。对于内相,将90g的油相置于烧杯中,并与7.78g的desmoduri(ipdi)和3.35g的mondurmr(mdi)混合,使用搅拌器混合直至均质。在反应器中,通过向144g的水中加入6g的丙烯酸酯阴离子聚合物(上述制备的),并用设定7℃的再循环水浴混合30分钟来开始制备水相。在混合水相之后,经1分钟,将内相加入到反应器中,并设定caframo的速度为2500rpm开始研磨,形成目标粒度(即10um)的稳定乳液。在研磨结束时,关掉搅拌器,并用z-棒代替研磨叶片。然后,将搅拌速度设定为200rpm,并将该批料在120分钟内加热至60℃,保持在60℃240分钟,在60分钟内加热至85℃,并保持在85℃240分钟。在加热循环完成之后,将温度设定为恢复室温。实施例2b:使用caframobdc6015搅拌器、finemechkgw-2205回火烧杯(反应器)和coleparmer再循环加热水浴制备该批料。对于内相,将90g的油相置于烧杯中,并与7.78g的desmoduri(ipdi)和3.35g的mondurmr(mdi)混合,使用搅拌器混合直至均质。在反应器中,通过向144g的水中加入6g的丙烯酸酯阴离子聚合物(上述制备的),并用设定7℃的再循环水浴混合30分钟来开始制备水相。在混合水相之后,经1分钟,将内相加入到反应器中,并设定caframo的速度为2500rpm开始研磨,形成目标粒度(即10um)的稳定乳液。在研磨结束时,关掉搅拌器,并用z-棒代替研磨叶片。然后,将搅拌速度设定为200rpm,并将该批料在120分钟内加热至60℃,保持在60℃下120分钟,并加入1.8g另外的交联剂(deta)。将该批料在60℃下继续加热120分钟,在60分钟内将批料加热至85℃,并保持在85℃240分钟。在加热循环完成之后,将温度设定为恢复室温。微胶囊包含具有表面电荷官能团的聚丙烯酸酯/聚脲/聚氨酯三组分壁。该微胶囊是阴离子的,并且显示出低渗漏。实施例3聚合物的制备方法∶对于水相1,将0.5克水溶性的引发剂(v50)加入到在40℃下夹套钢制反应器中的200g水中,同时以1000rpm混合,并且以100cc/min用氮气覆盖。在45分钟内,将溶液从40℃加热至75℃,保持在75℃45分钟,并且在75分钟内冷却至60℃。加入150g的水相2,其包括5g的至少一种多官能水分散性丙烯酸单体(sr415)和5.5克的至少一种具有交联官能团的水溶性丙烯酸单体(即,tbaema),并且混合提高至1500rpm。在60℃下,将合并的水相混合60分钟,然后加入150g的水相3,其包括12.5克的至少一种具有表面电荷官能团的水溶性丙烯酸单体(即,2-(甲基丙烯酰氧基)乙基]三甲基氯化铵),并将合并的溶液保持在60℃下另外30分钟。然后,在30分钟内将溶液加热到75℃,在7小时内从75℃加热至95℃,并保持在95℃下6小时,形成最终聚合物。实施例3a:使用caframobdc6015搅拌器、finemechkgw-2205回火烧杯(反应器)和coleparmer再循环加热水浴制备该批料。对于内相,将90g的油相置于烧杯中,并与7.78g的desmoduri(ipdi)和3.35g的mondurmr(mdi)混合,使用搅拌器混合直至均质。在反应器中,通过向144g的水中加入6g的丙烯酸酯阳离子聚合物(上述制备的),并用设定7℃的再循环水浴混合30分钟来开始制备水相。在混合水相之后,经1分钟将内相加入到反应器中,并将caframo的速度设定为2500rpm开始研磨,形成目标粒度(即,10um)的稳定乳液。在研磨结束时,关掉搅拌器,并用z-棒代替研磨叶片。然后,将搅拌速度设定为200rpm,并将该批料在120分钟内加热至60℃,保持在60℃240分钟,在60分钟内加热至85℃,并保持在85℃240分钟。在加热循环完成之后,将温度设定为恢复室温。实施例3b:使用caframobdc6015搅拌器、finemechkgw-2205回火烧杯(反应器)和coleparmer再循环加热水浴制备该批料。对于内相,将90g的油相置于烧杯中,并与7.78g的desmoduri(ipdi)和mondurmr(mdi)混合,使用搅拌器混合直至均质。在反应器中,通过向144g的水中加入6g的丙烯酸酯阳离子聚合物(上述制备的),并用设定7℃的再循环水浴混合30分钟来开始制备水相。在混合水相之后,经1分钟将内相加入到反应器中,并将caframo的速度设定为2500rpm开始研磨,形成目标粒度(即,10um)的稳定乳液。在研磨结束时,关掉搅拌器,并用z-棒代替研磨叶片。然后,将搅拌速度设定为200rpm,并将该批料在120分钟内加热至60℃,保持在60℃120分钟,并加入1.8g另外的交联剂(deta)。将该批料在60℃下继续加热120分钟,在60分钟内将批料加热至85℃,并保持在85℃240分钟。在加热循环完成之后,将温度设定为恢复室温。微胶囊包含具有带表面电荷官能团的聚丙烯酸酯/聚脲双组分壁系统。该胶囊是阳离子型的,并且显示出低渗漏。实施例4聚合物的制备方法∶对于水相1,将0.5克水溶性的引发剂(v50)加入到在40℃下夹套钢制反应器中的200g水中,同时以1000rpm混合,并且以100cc/min用氮气覆盖。在45分钟内,将溶液从40℃加热至75℃,保持在75℃45分钟,并且在75分钟内冷却至60℃。加入150g的水相2,其包括2.5g的至少一种多官能水分散性丙烯酸单体(sr415)和10克的至少一种具有交联官能团的水溶性丙烯酸单体(即tbaema),并且混合提高至1500rpm。在60℃下,将合并的水相混合60分钟,然后加入150g的水相3,其包括12.5克的至少一种具有表面电荷官能团的水溶性丙烯酸单体(2-磺乙基甲基丙烯酸酯),同时调节ph为6.0,并将合并的溶液保持在60℃另外30分钟。然后,在30分钟内将溶液加热到75℃,在7小时内从75℃加热至95℃,并保持在该温度下6小时,形成最终聚合物。实施例4:使用caframobdc6015搅拌器、finemechkgw-2205回火烧杯(反应器)和coleparmer再循环加热水浴制备该批料。对于内相,将90g的油相置于烧杯中,并与7.78g的desmoduri(ipdi)和3.35g的mondurmr(mdi)混合,使用搅拌器混合直至均质。在反应器中,通过向144g的水中加入6g的丙烯酸酯阴离子聚合物(上述制备的),并用设定7℃的再循环水浴混合30分钟来开始制备水相。在混合水相之后,经1分钟,将内相加入到反应器中,并设定caframo的速度为2500rpm开始研磨,形成目标粒度(即10um)的稳定乳液。在研磨结束时,关掉搅拌器,并用z-棒代替研磨叶片。然后,将搅拌速度设定为200rpm,并将该批料在120分钟内加热至60℃,保持在60℃240分钟,在60分钟内加热至85℃,并保持在85℃240分钟。在加热循环之后,将温度设定为恢复室温。所有样品的最后包封都可以包含具有表面电荷官能团的聚丙烯酸酯/聚脲双组分壁系统。该微胶囊是ph非依赖性阴离子的,并且显示出低渗漏。实施例5聚合物的制备方法∶对于水相1,将0.5克水溶性的引发剂(v50)加入到在40℃下夹套钢制反应器中的200g水中,同时以1000rpm混合,并且以100cc/min用氮气覆盖。将溶液在45分钟内从40℃加热至75℃,保持在75℃45分钟,并在75分钟内冷却至60℃。加入150g的水相2,其包括2.5g的至少一种多官能水分散性丙烯酸单体(乙氧基化的三羟甲基丙烷三丙烯酸酯,比如sr9035或sr502)和10克的至少一种具有交联官能团的水溶性丙烯酸单体(即tbaema),并且混合提高至1500rpm。在60℃下,将合并的水相混合60分钟,然后加入150g的水相3,其包括12.5克的至少一种具有表面电荷官能团的水溶性丙烯酸单体(cd9055),并将合并的溶液保持在60℃另外30分钟。然后,在30分钟内将溶液加热到75℃,在7小时内从75℃加热至95℃,并保持在该温度下6小时,形成最终聚合物。实施例5a:使用caframobdc6015搅拌器、finemechkgw-2205回火烧杯(反应器)和coleparmer再循环加热水浴制备该批料。对于内相,将90g的油相置于烧杯中,并与7.78g的desmoduri(ipdi)和3.35g的mondurmr(mdi)混合,使用搅拌器混合直至均质。在反应器中,通过向144g的水中加入6g的丙烯酸酯阴离子聚合物(上述制备的),并用设定7℃的再循环水浴混合30分钟来开始制备水相。在混合水相之后,经1分钟,将内相加入到反应器中,并设定caframo的速度为2500rpm开始研磨,形成目标粒度(即10um)的稳定乳液。在研磨结束时,关掉搅拌器,并用z-棒代替研磨叶片。然后,将搅拌速度设定为200rpm,并将该批料在120分钟内加热至60℃,保持在60℃240分钟,在60分钟内加热至85℃,并保持在85℃240分钟。在加热循环之后,将温度设定为恢复室温。实施例5b:使用caframobdc6015搅拌器、finemechkgw-2205回火烧杯(反应器)和coleparmer再循环加热水浴制备该批料。对于内相,将90g的油相置于烧杯中,并与7.78g的desmoduri(ipdi)和3.35g的mondurmr(mdi)混合,使用搅拌器混合直至均质。在反应器中,通过向144g的水中加入6g的丙烯酸酯阴离子聚合物(上述制备的),并用设定7℃的再循环水浴混合30分钟来开始制备水相。在混合水相之后,经1分钟,将内相加入到反应器中,并设定caframo的速度为2500rpm开始研磨,形成目标粒度(即10um)的稳定乳液。在研磨结束时,关掉搅拌器,并用z-棒代替研磨叶片。然后,将搅拌速度设定为200rpm,并将该批料在120分钟内加热至60℃,保持在60℃另外20分钟,并加入1.8g另外的交联剂(deta)。然后,将该批料保持在60℃另外120分钟,在60分钟内加热至85℃,并保持在85℃240分钟。在加热循环完成之后,将温度设定为恢复室温。所有样品的最后包封都可以包含具有表面电荷官能团的聚丙烯酸酯/聚脲双组分壁系统。该微胶囊是阴离子的,并且显示出低渗漏。实施例6聚合物的制备方法∶对于水相1,将0.5克水溶性的引发剂(v50)加入到在40℃下夹套钢制反应器中的200g水中,同时以1000rpm混合,并且以100cc/min用氮气覆盖。将溶液在45分钟内从40℃加热至75℃,保持在75℃45分钟,并在75分钟内冷却至60℃。加入150g的水相2,其包括2.5g的至少一种多官能水分散性丙烯酸单体(二丙烯酸聚乙二醇酯,比如sr344或sr601)和10克的至少一种具有交联官能团的水溶性丙烯酸单体(即tbaema),并且混合提高至1500rpm。在60℃下,将合并的水相混合60分钟,然后加入150g的水相3,其包括12.5克的至少一种具有表面电荷官能团的水溶性丙烯酸单体(cd9055),并将合并的溶液保持在60℃另外30分钟。然后,在30分钟内将溶液加热到75℃,在7小时内从75℃加热至95℃,并保持在该温度下6小时,形成最终聚合物。实施例6a:使用caframobdc6015搅拌器、finemechkgw-2205回火烧杯(反应器)和coleparmer再循环加热水浴制备该批料。对于内相,将90g的油相置于烧杯中,并与7.78g的desmoduri(ipdi)和3.35g的mondurmr(mdi)混合,使用搅拌器混合直至均质。在反应器中,通过向144g的水中加入6g的丙烯酸酯阴离子聚合物(上述制备的),并用设定7℃的再循环水浴混合30分钟来开始制备水相。在混合水相之后,经1分钟,将内相加入到反应器中,并设定caframo的速度为2500rpm开始研磨,形成目标粒度(即10um)的稳定乳液。在研磨结束时,关掉搅拌器,并用z-棒代替研磨叶片。然后,将搅拌速度设定为200rpm,并将该批料在120分钟内加热至60℃,保持在60℃240分钟,在60分钟内加热至85℃,并保持在85℃240分钟。在加热循环之后,将温度设定为恢复室温。实施例6b:使用caframobdc6015搅拌器、finemechkgw-2205回火烧杯(反应器)和coleparmer再循环加热水浴制备该批料。对于内相,将90g的油相置于烧杯中,并与7.78g的desmoduri(ipdi)和3.35g的mondurmr(mdi)混合,使用搅拌器混合直至均质。在反应器中,通过向144g的水中加入6g的丙烯酸酯阴离子聚合物(上述制备的),并用设定7℃的再循环水浴混合30分钟来开始制备水相。在混合水相之后,经1分钟,将内相加入到反应器中,并设定caframo的速度为2500rpm开始研磨,形成目标粒度(即10um)的稳定乳液。在研磨结束时,关掉搅拌器,并用z-棒代替研磨叶片。然后,将搅拌速度设定为200rpm,并将该批料在120分钟内加热至60℃,保持在60℃另外20分钟,并加入1.8g另外的交联剂(deta)。然后,将该批料保持在60℃另外120分钟,在60分钟内加热至85℃,并保持在85℃240分钟。在加热循环完成之后,将温度设定为恢复室温。所有样品的最后包封都可以包含具有表面电荷官能团的聚丙烯酸酯/聚脲双组分壁系统。该微胶囊是ph非依赖性阴离子的,并且显示出低渗漏。实施例7聚合物的制备方法∶对于水相1,将0.5克水溶性的引发剂(v50)加入到在40℃下夹套钢制反应器中的200g水中,同时以1000rpm混合,并且以100cc/min用氮气覆盖。在45分钟内,将溶液从40℃加热至75℃,保持在75℃45分钟,并且在75分钟内冷却至60℃。加入150g的水相2,其包括2.5g的至少一种多官能水分散性丙烯酸单体(二甲基丙烯酸聚乙二醇酯,比如sr603)和10克的至少一种具有交联官能团的水溶性丙烯酸单体(即tbaema),并且混合提高至1500rpm。在60℃下,将合并的水相混合60分钟,然后加入150g的水相3,其包括12.5克的至少一种具有表面电荷官能团的水溶性丙烯酸单体(cd9055),并将合并的溶液保持在60℃另外30分钟。然后,在30分钟内将溶液加热到75℃,在7小时内从75℃加热至95℃,并保持在该温度下6小时,形成最终聚合物。实施例7a:使用caframobdc6015搅拌器、finemechkgw-2205回火烧杯(反应器)和coleparmer再循环加热水浴制备该批料。对于内相,将90g的油相置于烧杯中,并与7.78g的desmoduri(ipdi)和3.35g的mondurmr(mdi)混合,使用搅拌器混合直至均质。在反应器中,通过向144g的水中加入6g的丙烯酸酯阴离子聚合物(上述制备的),并用设定7℃的再循环水浴混合30分钟来开始制备水相。在混合水相之后,经1分钟,将内相加入到反应器中,并设定caframo的速度为2500rpm开始研磨,形成目标粒度(即10um)的稳定乳液。在研磨结束时,关掉搅拌器,并用z-棒代替研磨叶片。然后,将搅拌速度设定为200rpm,并将该批料在120分钟内加热至60℃,保持在60℃240分钟,在60分钟内加热至85℃,并保持在85℃240分钟。在加热循环之后,将温度设定为恢复室温。实施例7b:使用caframobdc6015搅拌器、finemechkgw-2205回火烧杯(反应器)和coleparmer再循环加热水浴制备该批料。对于内相,将90g的油相置于烧杯中,并与7.78g的desmoduri(ipdi)和3.35g的mondurmr(mdi)混合,使用搅拌器混合直至均质。在反应器中,通过向144g的水中加入6g的丙烯酸酯阴离子聚合物(上述制备的),并用设定7℃的再循环水浴混合30分钟来开始制备水相。在混合水相之后,经1分钟,将内相加入到反应器中,并设定caframo的速度为2500rpm开始研磨,形成目标粒度(即10um)的稳定乳液。在研磨结束时,关掉搅拌器,并用z-棒代替研磨叶片。然后,将搅拌速度设定为200rpm,并将该批料在120分钟内加热至60℃,保持在60℃另外20分钟,并加入1.8g另外的交联剂(deta)。然后,将该批料保持在60℃另外120分钟,在60分钟内加热至85℃,并保持在85℃240分钟。在加热循环完成之后,将温度设定为恢复室温。所有样品的最后包封都可以包含具有表面电荷官能团的聚丙烯酸酯/聚脲双组分壁系统。该微胶囊是ph非依赖性阴离子的,并且显示出低渗漏。实施例8:微胶囊性质的表征。微胶囊悬浮液中游离油的表征:通过使用自动体积分配器,将1g的微胶囊悬浮液(40%固体)与10ml的己烷/dbp溶液混合,以从微胶囊悬浮液浸析出游离油,然后放置在台子上30分钟。小心地用移液管吸取1ml上面澄清的己烷/dbp层,并通过agilent6890n气相色谱法(gc)测量来测定悬浮液中的游离油。将游离油的结果显示在下表1中:表1样品1a1b1c2a2b3a3b游离油(%)0.070.030.060.110.020.070.03所有测试样品的游离油低表明这是一种成功的微囊包封方法,其可以高效地包封核芯物质,具有极低渗漏。在己烷中微胶囊的核芯渗漏的表征:在150ml广口瓶中,将微胶囊悬浮液(包括1.5g核芯物质)与47ml的去离子水混合,形成均质悬浮液。将50ml的己烷w/dbp轻轻地加入到所述广口瓶中,并盖紧盖子。在t=24小时、1周、2周和4周,小心地用移液管移取上面己烷层,并在不同时间点,通过agilent6890ngas色谱法(gc)测量萃取物来测定微胶囊悬浮液的渗漏。将渗漏结果显示在图1中。在己烷中长期渗漏(至多4周)的结果显示出这些微胶囊在有机溶剂中可非常稳定,特别是含有另外的交联剂(deta)的这些样品。结果指示多组分壁系统对于有机溶剂系统具有高度抗性。微胶囊样品的表面电荷的表征∶将10g的微胶囊水性悬浮液(4%固体)加入到清洗干净的样品杯中,并将ph用0.1nnaoh调节至10。通过使用0.1nhcl(10ul/min)慢慢地调节水性悬浮液的ph从10至3,并通过microtracstabinoparticlechargetitrationanalyzer测量微胶囊样品的表面电荷,显示在图2、3、4和5中。试验结果显示微胶囊样品的表面区域上具有永久电荷,更重要的是,可以通过使用具有电荷官能团的不同丙烯酸单体定制表面电荷。由于来自cd9055的羧基,样品1和2都具有ph-依赖性阴离子表面电荷,由于来自2-磺乙基甲基丙烯酸酯的硫酸根基团,样品4具有ph-非依赖性阴离子表面电荷,而样品3的阳离子表面电荷来自2-(甲基丙烯酰氧基)乙基]三甲基氯化铵的三甲基铵基团。将本文说明书中引用的所有文件的相应部分都通过援引其中这样的并入被允许的所有范围并入本文中。引用的所有出版物都用于其在申请日之前的公开内容,而不应当被看作是这样的出版物属于现有技术或由于在先发明而使本发明没有资格享有这样的公开。对于本文中术语的任何含义或定义与通过援引并入的文件的相同术语的任何含义或定义不一致的情形,则以本文中赋予该术语的含义或定义为准。本文公开的尺寸和值不应当理解为严格地限制于列举的精确数值。相反,除非另有说明,每个这样的尺寸都意味着涵盖所列举的值和该值周围的功能等效范围。例如,公开为“40mm”的尺寸意味着指约“40mm”。使用单数术语比如“一个”、“一种”旨在涵盖单数和复数,除非本文另有说明或明显与本文内容矛盾。术语包含“具有”、“包括”和“含有”应当被看作是开放性术语。作为“优选的”实施方案的某些实施方案以及作为优选的实施方案、特征或范围的其他列举或提出其是优选的的任何说明都不视为是限制性的。除非本文另有说明或者明显与本文内容矛盾,否则本文描述的所有方法都可以以任何合适的顺序进行。本文提供的任何或所有的实施例、或示例性术语(例如“比如”)的使用旨在阐述本发明,而不造成对本发明范围的限制。未声明的术语不应当被认为限制本发明的范围。除非反映在附加权利要求书中,否则本文中关于某些特征构成所主张的发明的组分的任何声明或建议都不意味着限制。上述说明书中已经描述了本发明的原理、优选的实施方案和操作模式。然而,本文中旨在保护的本发明不应当被看作是限制所公开的特定形式,因为这些被认为是示例性的而不是限制性的,在没有背离本发明的精神和范围下,本领域技术人员可以进行多种变化和改变。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1