一种从植物中定向分离目标化合物的方法与流程
本发明属于植物化学及其分析化学技术领域,具体地说,涉及一种从植物中定向分离目标化合物的方法。
背景技术:
标准品,即标准物品,是中药标准对照品研究中心代理的作为一种衡量标准;用做药物方面,则为含量测定中的标准含量。标准品包括化学计量标准品、冶金标准品和药检标准品。
标准品是国家药品标准中用于鉴别、检查、含量测定、杂质和有关物质检查等标准物质,它是国家药品标准不可分割的组成部分。国家药品标准物质是国家药品标准的物质基础,它是用来检查药品质量的一种特殊的专用量具;是测量药品质量的基准;也是作为校正测试仪器与方法的物质标准。在药品检验中,它是确定药品真伪优劣的对照,是控制药品质量必不可少的工具。
测量是科学的基础,而具有准确量值的“标准物质”是定性定量测试分析的“标杆”。自动化和标准化的现代工业生产配套设备及生产过程都离不开标准物质的质量保证和质量控制,各种国际和国家标准的实施和推广,也需要标准物质来保证测量结果的可靠性和有效性。
根据“国际通用计量学基本术语”和“国际标准化组织(internationalorganizationforstandardization,iso)指南30”的定义,标准物质(referencematerial,rm)是具有一种或多种足够均匀和很好地确定了的特性值,用以校准设备,评价测量方法或给材料赋值的材料或物质;而有证标准物质(certifiedreferencematerial,crm):是附有证书的标准物质,其一种或多种特性值用建立了溯源性的程序确定,使之可溯源到准确复现的用于表示该特性值的计量单位,每个标准值都附有给定置信水平的不确定度。标准物质的特性量值具有均匀性、稳定性、准确性和复现性的特点。它的应用已遍及工业生产、商业贸易、环境保护和医疗卫生等诸多领域,在产品质量保证、仲裁检验、分析测试技术发展、计量学发展等方面发挥重要的作用。可以说:有需要分析测量的领域就存在标准物质的需求。因此,标准物质的发展对科技的发展意义重大。
技术实现要素:
有鉴于此,本发明提供了一种从植物中定向分离目标化合物的方法。
为了解决上述技术问题,本发明公开了一种从植物中定向分离目标化合物的方法,包括如下步骤:
步骤1、植物的浸提和浓缩:
对植物样品自然晾干,经粉碎机粉碎并过筛,样品粉末用重量比2~3倍的95%乙醇/水溶液浸泡,超声热提,共提取4~6次,每次2小时,过滤、合并提取液,水浴减压浓缩,得到样品浸膏;
步骤2、高效液相色谱法按相同时间段精确分段:
将浸膏用总量比3倍量的乙腈超声温水浴中溶解,待其溶解后用0.22μm滤膜过滤,得滤液,滤液采用高效液相色谱法梯度洗脱进行分离,分离样品液按相同时间段收集进行精确分段,按时间段顺序编号;
步骤3、质谱筛分目标化合物所在段位:
将上述步骤2中按相同时间段收集进行精确分段的样品,进行质谱检测分析,后通过检索图谱找出目标化合物所在的段位;且重复步骤2及其3多次,以最简要的段位进行下一步骤的分离纯化;
步骤4、分离纯化目标化合物:
对上述步骤3中目标化合物所在的段位,进行高效液相色谱法分离纯化;收集每个色谱信号峰,记录每个信号峰最后的重量,使其足够用于波谱分析检测的量,通过波谱数据鉴定其目标化合物结构。
可选地,步骤1中的筛目数为80目;过筛后的植物样品水分要求<5wt%。
可选地,步骤1中的超声热提的温度47~53℃,超声功率为200~300w;水浴温度53~58℃,真空压强为0.06~0.08mpa。
可选地,步骤2中的水浴温度为47~53℃。
可选地,步骤2中的高效液相色谱法的梯度洗脱极性为10%的乙腈/水溶液增至纯乙腈,时间总量程为80分钟。
可选地,步骤2中的所述的相同时间段为均分时间总量程的每一个单元时间段。
可选地,步骤3中的质谱条件,包括:离子源、电离能、离子源温度、传输线温度、检测模式、质量扫描范围和溶剂延迟。
可选地,步骤4中的纯化最终纯度为:95±3%;分离纯化条件为:90%乙腈水溶液等度洗脱。
可选地,步骤4中的用于波谱分析检测的量至少为5±3mg。
可选地,步骤4中的波谱分析包括:质谱、核磁共振普;波谱数据包括:质谱、核磁共振谱。
与现有技术相比,本发明可以获得包括以下技术效果:
1)本发明的方法无需标准品,仅需一定量的植物样品;
2)本发明寻找植物中目标化合物时,样品及其试剂用量少;
3)本发明实验周期短;实验过程更加的快捷、方便和经济。
当然,实施本发明的任一产品并不一定需要同时达到以上所述的所有技术效果。
附图说明
此处所说明的附图用来提供对本发明的进一步理解,构成本发明的一部分,本发明的示意性实施例及其说明用于解释本发明,并不构成对本发明的不当限定。在附图中:
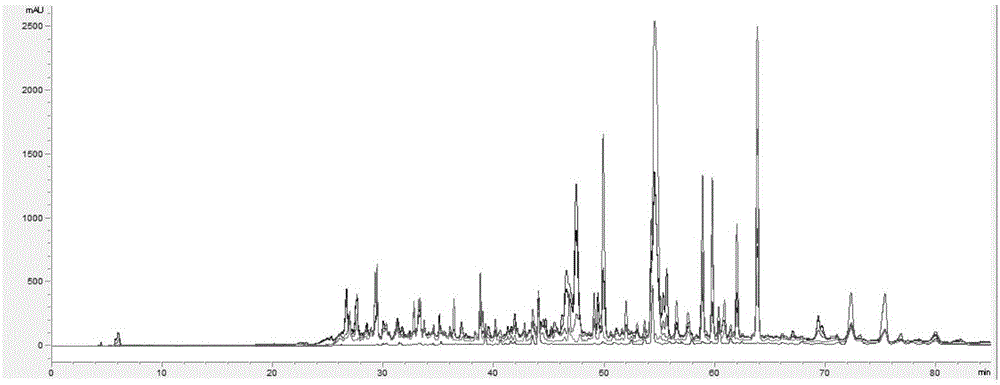
图1为植物样品的高效液相色谱法(hplc)梯度洗脱分析图谱;
图2为0~10min单元时间段液相分离总化合物的总离子流色谱图;
图3为10~20min单元时间段液相分离总化合物的总离子流色谱图;
图4为20~30min单元时间段液相分离总化合物的总离子流色谱图;
图5为30~40min单元时间段液相分离总化合物的总离子流色谱图;
图6为40~50min单元时间段液相分离总化合物的总离子流色谱图;
图7为50~60min单元时间段液相分离总化合物的总离子流色谱图;
图8为60~70min单元时间段液相分离总化合物的总离子流色谱图;
图9为70~80min单元时间段液相分离总化合物的总离子流色谱图;
图10为40~50min单元时间段收集混合样品液的高效液相色谱法(hplc)等度洗脱分析图谱;
图11为第2个主色谱信号峰的1h-nmr;
图12为第2个主色谱信号峰的13c-nmr;
图13为大麻二酚的结构式。
具体实施方式
以下将配合实施例来详细说明本发明的实施方式,藉此对本发明如何应用技术手段来解决技术问题并达成技术功效的实现过程能充分理解并据以实施。
本发明公开了一种从植物中定向分离目标化合物的方法,包括如下步骤:
步骤1、植物的浸提和浓缩:
对植物样品自然晾干,经粉碎机粉碎过(80目筛),样品粉末用重量比2~3倍的95%乙醇/水溶液浸泡,超声热提,浸泡温度47~53℃,超声功率为200~300w,共提取4~6次,每次2小时,过滤、合并提取液,水浴减压浓缩,水浴温度53~58℃,真空压强为0.06~0.08mpa,得到样品浸膏。
步骤2、高效液相色谱法(hplc)按相同时间段精确分段:
将浸膏用总量比3倍量的乙腈超声温水浴中溶解,水浴温度为47~53℃,待其溶解后用0.22μm滤膜过滤,得滤液,滤液使用高效液相色谱法(hplc)梯度洗脱进行分离,分离样品液按相同时间段收集进行精确分段,洗脱剂从10%的乙腈/水溶液增至纯乙腈,时间总量程为80分钟;按每10min为一时间段顺序编号。
步骤3、质谱筛分目标化合物所在段位
将上述步骤2中按相同时间段收集进行精确分段的样品,进行质谱检测分析,后通过检索图谱找出目标化合物所在的段位。且可重复步骤2及其3多次,以最简要的段位进行下一步骤的分离纯化。
其中,质谱条件包括:离子源、电离能、离子源温度、传输线温度、检测模式、质量扫描范围和溶剂延迟,检测条件如下:
agilent5977gc/msd气质联用仪;
气相色谱分析条件:色谱柱为hp-5ms毛细管色谱柱(30m×0.25mm×0.25μm),进样口温度为280℃;程序升温:初始温度50℃,保持2min,以10℃/min升至100℃,保持2min,以10℃/min升至150℃,保持2min,以10℃/min升至200℃,保持20min,以10℃/min升至280℃,保持3min。载气:高纯氦气,流速20ml/min;进样方式:不分流;进样体积:2.0μl。
质谱条件:离子源:ei源;电离能:70ev;离子源温度:230℃;传输线温度:300℃;检测模式:全离子扫描监测,质量扫描范围:35~600amu;溶剂延迟:2min。
步骤4、分离纯化目标化合物
对上述步骤3中目标化合物所在的时间段位,采用高效液相色谱法(hplc)分离纯化,分离纯化条件为:90%乙腈水溶液等度洗脱;收集每个色谱信号峰,记录每个信号峰最后的重量,使其足够用于波谱分析检测的量,通过波谱数据鉴定其目标化合物结构。
本发明采用高效液相色谱法(hplc)定向分离目标化合物,检测条件如下:
高效液相色谱仪agilent1260(多波长检测器mwd,二元泵),色谱柱:shimadzushim-packgls,10mm×250mm长,i.d.c18柱,5μm粒度;柱温:25℃;流动相:乙腈:水=9:1;流速:3.0ml/min;进样量:20μl;检测波长:多波长检测(主波长为230nm)。
实施例1从工业大麻植物中分离提取目标化合物大麻二酚:
1.1植物的浸提和浓缩
称取50.1g工业大麻的叶,经粉碎机粉碎,样品粉末过80目筛筛分,将工业大麻样品粉末用重量比2倍量的95%乙醇/水溶液浸泡,超声热浸提取,浸泡温度50℃,超声功率为250w,共提取4次,每次2小时;过滤、合并提取液,水浴减压浓缩蒸去乙醇/水溶液,水浴温度55℃,真空压强为0.07mpa,得到4.3g样品浸膏。
1.2高效液相色谱法(hplc)按相同时间段精确分段
将浸膏用总量比3倍量的乙腈超声温水浴中溶解,水浴温度为50℃,待其溶解后用0.22μm滤膜过滤,滤液用高效液相色谱法(hplc)梯度洗脱,洗脱剂从10%的乙腈/水溶液增至纯乙腈,时间总量程为80分钟。按此技术要求分离,分离得到的样品液按每10min为一个单元时间段收集而进行精确分段,并对每个单元时间段按顺序编号,其植物样品采用高效液相色谱法(hplc)梯度洗脱分析图谱参见图1。
1.3质谱筛分目标化合物所在段位
将上述步骤1.2中按每10min为一个单元时间段收集而进行精确分段的样品,进行质谱检测分析,其每个时间段的液相分离总化合物总离子流色谱图参见图2~9,通过检索图谱找出目标化合物所在的段位为40-50min单元时间段,该时间段的液相分离总化合物总离子流色谱图参见图6。
1.4分离纯化目标化合物
对上述步骤1.3中得出的目标化合物所在的段位为40-50min单元时间段,采用高效液相色谱法(hplc)分离纯化,分离纯化条件为:90%乙腈水溶液等度洗脱。收集该单元时间段里的2个主色谱信号峰,收集到第1个主色谱信号峰最后的重量为15.5mg,第2个主色谱信号峰最后的重量为10.7mg,通过波谱数据鉴定2个信号峰的化学结构。
其40~50min单元时间段收集的混合样品液经高效液相色谱法(hplc)等度洗脱分析图谱参见图10。
样品经过核磁共振波谱分析,确认第2个主色谱信号峰为大麻二酚,其1h-nmr参见图11,13c-nmr图谱参见图12,大麻二酚的结构式参见图13。
实施例2从工业大麻植物中分离提取目标化合物大麻二酚:
1.1植物的浸提和浓缩
称取60.6g工业大麻的籽,经粉碎机粉碎,样品粉末过80目筛筛分,将工业大麻样品粉末用重量比2倍量的95%乙醇/水溶液浸泡,超声热浸提取,水浴温度47℃,超声功率为300w,共提取4次,每次2小时;过滤、合并提取液,水浴减压浓缩蒸去乙醇/水溶液,水浴温度53℃,真空压强为0.08mpa,得到5.1g样品浸膏。
1.2高效液相色谱法(hplc)按相同时间段精确分段
将浸膏用总量比3倍量的乙腈超声温水浴中溶解,水浴温度为47℃,待其溶解后用0.22μm滤膜过滤,滤液用高效液相色谱法(hplc)梯度洗脱,洗脱剂从10%的乙腈/水溶液增至纯乙腈,时间总量程为80分钟。按此技术要求分离,分离得到的样品液按每10min为一个单元时间段收集而进行精确分段,并对每个单元时间段按顺序编号。
1.3质谱筛分目标化合物所在段位
将上述步骤1.2中按每10min为一个单元时间段收集而进行精确分段的样品,进行质谱检测分析,后通过检索图谱找出目标化合物所在的段位为40-50min单元时间段。
1.4分离纯化目标化合物
对上述步骤1.3中得出的目标化合物所在的段位为40-50min单元时间段,采用高效液相色谱法(hplc)分离纯化,分离纯化条件为:90%乙腈水溶液等度洗脱。收集该单元时间段里的2个主色谱信号峰,收集到第1个主色谱信号峰最后的重量为17.2mg,第2个主色谱信号峰最后的重量为12.0mg,通过核磁波谱数据鉴定2个信号峰的化学结构。
实施例3从工业大麻植物中分离提取目标化合物大麻二酚:
称取70.4g工业大麻的籽,经粉碎机粉碎,样品粉末过80目筛筛分,将工业大麻样品粉末用重量比3倍量的95%乙醇/水溶液浸泡,超声热浸提取,水浴温度53℃,超声功率为200w,共提取4次,每次2小时;过滤、合并提取液,水浴减压浓缩蒸去乙醇/水溶液,水浴温度58℃,真空压强为0.06mpa,得到5.8g样品浸膏。
1.2高效液相色谱法(hplc)按相同时间段精确分段
将浸膏用总量比3倍量的乙腈超声温水浴中溶解,水浴温度为53℃,待其溶解后用0.22μm滤膜过滤,滤液用高效液相色谱法(hplc)梯度洗脱,洗脱剂从10%的乙腈/水溶液增至纯乙腈,时间总量程为80分钟。按此技术要求分离,分离得到的样品液按每10min为一个单元时间段收集而进行精确分段,并对每个单元时间段按顺序编号。
1.3质谱筛分目标化合物所在段位
将上述步骤1.2中按每10min为一个单元时间段收集而进行精确分段的样品,进行质谱检测分析,后通过检索图谱找出目标化合物所在的段位为40-50min单元时间段。
1.4分离纯化目标化合物
对上述步骤1.3中得出的目标化合物所在的段位为40-50min单元时间段,采用高效液相色谱法(hplc)分离纯化,分离纯化条件为:90%乙腈水溶液等度洗脱。收集该单元时间段里的2个主色谱信号峰,收集到第1个主色谱信号峰最后的重量为20.1mg,第2个主色谱信号峰最后的重量为15.4mg,通过核磁波谱数据鉴定2个信号峰的化学结构。
上述说明示出并描述了发明的若干优选实施例,但如前所述,应当理解发明并非局限于本文所披露的形式,不应看作是对其他实施例的排除,而可用于各种其他组合、修改和环境,并能够在本文所述发明构想范围内,通过上述教导或相关领域的技术或知识进行改动。而本领域人员所进行的改动和变化不脱离发明的精神和范围,则都应在发明所附权利要求的保护范围内。
- 还没有人留言评论。精彩留言会获得点赞!