一种合成气油-醇联产的催化剂及其制备与应用
1.本发明属于工业催化剂开发技术领域,具体涉及一种合成气油-醇联产的催化剂及其制备与应用。更具体地,该催化剂的一般组成形式为co-x-y-z/ac,是一种以活性炭为载体的负载型钴基催化剂,辅助添加zr、zn、cu、cr中的一种或二种以上为第一助剂,mn、la、ce、mo、v中的一种或二种以上为第二助剂,li、na、k、mg、ca、sr中的一种或二种以上为第三助剂以提高催化剂活性、产物中烯烃和含氧化合物的选择性并延长催化剂的使用寿命,并结合烯烃氢甲酰化以及加氢等技术,实现以合成气为原料的油-醇联产过程。
背景技术:
2.我国石油资源相对匮乏,而煤炭、天然气等碳质资源相对丰富,清洁高效地利用煤炭等资源可以在一定程度上缓解我国的能源短缺和环境污染等问题。多种碳质资源可以利用不同的技术手段转化为合成气,从合成气出发经费托合成过程直接制备清洁液体燃料以及烯烃、含氧化合物等高值化学品的油-醇联产新化工生产过程逐渐成为未来碳一化学领域的研究重点。
3.烯烃是一类非常重要的大宗化学品,每年的消耗量十分巨大。烯烃包括低碳烯烃(c2~c4)和高碳烯烃(c
5+
)。具体来说,c2~c4的低碳烯烃是化学工业的重要基本原料,可用于生产聚乙烯、聚丙烯等化学品,具有广阔的市场空间。碳数高于5的烯烃可以通过氢甲酰化技术等手段,转化为相应的醛、醇、酸、卤化物等高附加值精细化学品。在石油化工中,低碳烯烃的生产主要来自于石脑油的裂解和流化催化裂化(fcc);在煤化工中,低碳烯烃主要来源于甲醇制烯烃(mto)。工业上高碳烯烃的来源主要是低碳烯烃齐聚。甲醇制烯烃是从合成气出发间接制备低碳烯烃的主要来源。该过程是指合成气先在铜基催化剂上制备甲醇,然后在分子筛的作用下将甲醇转化为低碳烯烃(主要是乙烯和丙烯)。合成气直接制备低碳烯烃一直是碳一化学的重要研究方向。从合成气出发直接制低碳烯烃,将会省去以甲醇或二甲醚为中间产物的过程,减少反应步骤,缩短工艺流程,在工业上能够极大地降低生产成本和设备投资,是极具价值的化工生产过程。但是,提高低碳烯烃在总产物中的选择性是一项重要挑战,也是工业化过程中的重要难题,很多研究者为此进行了大量的研究。孙予罕等人发现了一种暴露特定晶面的co2c纳米棱柱催化剂,该催化剂在温和的反应条件(523k和0.1~0.5mpa)下可实现合成气高选择性直接制备低碳烯烃,ch4选择性可低至5%,低碳烯烃选择性能够达到60%以上(不包括co2,~50%),同时烯/烷比大于30,产物分布不服从经典的asf分布规律,并且该催化剂具有良好的稳定性,反应600h仍未出现明显失活。通过深入的构效关系研究并结合dft理论计算,揭示了co2c存在显著的晶面效应,相比于其它暴露面,(101)和(020)晶面上ch4的生成势垒很高,能有效抑制ch4的形成,而(101)晶面非常有利于烯烃的生成。同时,他们又进一步研究了电子助剂na与共沉淀组分mn的作用。他们提出na向co转移电子,促进co吸附和解离,同时促进co2c物种的形成;在焙烧时形成的comn尖晶石型复合氧化物对co2c纳米棱柱的形成起到关键作用,而其它助剂(al、la、ce)只能促进球形co2c的形成。后续研究发现,随着反应压力的增加,发生菱形co2c向球形co2c的转变,同时产
生金属co,c2~c4低碳烯烃的选择性降低,含氧化合物的选择性增加。近期,他们通过研究不同载体负载的co2c催化剂发现,与sio2和al2o3载体相比,以cnts为载体更容易形成菱形co2c,烯烃和含氧化合物的选择性更高,这是因为co与碳载体的相互作用更弱。
4.从合成气出发直接制备高碳烯烃的报道较少,未见工业化报道。合成气制备高碳烯烃是一项重大挑战,因为在费托合成过程中调变烷烃和烯烃的选择性十分困难,抑制烯烃的加氢和二次反应成了该过程中的关键问题。马丁等人采用共沉淀法制备了一种zn、na改性的fe5c2催化剂并用于合成气直接制烯烃。其中,zn作为结构助剂,影响fe物种的尺寸;na作为电子助剂,富集在fe物种表面,促进co活化;催化剂表面的电子结构抑制了碳碳双键的加氢,促进了产物的脱附。fe-zn-0.36na催化剂在613k、2.0mpa、60000ml/h
·
g
cat
和h2/co/co2/ar=24/64/8/4的条件下,co转化率高达82.7%,c2~c4选择性为26.5%,c
5+
烃类选择性为35.9%,烯/烷比为6.3,c
5+
烯烃在烃类中的比例超过50%。孙予罕等人通过comn尖晶石制备得到的co2c基催化剂进行co加氢反应时,产物分布具有反asf分布的特殊性质,ch4的选择性远远低于asf分布的理论计算值,烃类产物主要分布于c2~c
12
之间,并且产物具有很高的烯烷比,烃类中烯烃的比例高达86%,为合成气直接制长链α-烯烃提供了新思路。
5.高碳醇(c
2+
oh)是指两个碳原子以上的直链脂肪族伯醇,分为低碳混合醇(c2~c5oh)和高碳混合醇(c
6+
oh),其在国民经济的多个领域中有着广泛的应用。低碳混合醇通常作为溶剂,也可以替代甲基叔丁基醚(mtbe)作为品质优良的汽油添加剂。高碳混合醇是合成增塑剂(c6~c9oh)、洗涤剂(c
10
~c
18
oh)、表面活性剂以及其它多种精细化学品的重要原料,在食品、医药、纺织、造纸等领域有广泛的应用。目前,工业上高碳脂肪醇的生产方法主要有两种,即齐格勒法和羰基合成法(oxo)。齐格勒法以乙烯为原料,以三乙基铝为催化剂实现碳链增长,再进行氧化、水解和分离工艺,制得c6~c
16
直链偶数伯醇。齐格勒法以三氧乙基铝(极易爆炸)为催化剂,且只能生产偶数碳原子的直链伯醇。而且,该方法工艺流程长,技术复杂,开发难度大,催化剂消耗量大且安全性差。羰基合成法是在钴基或铑基催化剂的作用下,烯烃与co和h2的混合气进行氢甲酰化反应生成醛,醛再加氢制得相应的醇。但是该法工艺流程长,催化剂价格昂贵且容易流失。但是,该方法的生产工艺复杂,产物与催化剂的分离以及催化剂的回收具有一定难度,限制了其大规模应用。据估算,高碳脂肪醇的市场售价在1万元/吨以上,年需求量约1300万吨,需求量年增长率约为3%。因此,开发工艺简单、反应条件温和、原料易得而且来源广泛的高碳脂肪醇生产新技术极具理论和实际意义。
6.通过co加氢反应以合成气为原料一步直接合成低碳混合醇(c1~c6oh),该法具有工艺简单的优点。专利us8048933公开了一种以合成气为原料生产低碳混合醇的方法,采用的催化剂为ni或na改性的mo2c基催化剂。专利us4752622和us4882630采用fe、co和ni助剂调变的mo、w基催化剂,并添加碱金属或碱土金属,用于合成气制低碳混合醇。us6753353公开了纳米mos2或w2c作为催化剂用于co加氢合成低碳混合醇。专利cn01130481在mos2基催化剂中引入mn元素,使得催化剂催化合成醇活性显著提高,同时提高c
2+
醇的选择性。专利cn101185895提供一种用于合成气合成低碳醇的催化剂及其制备方法,催化剂主要组分为cuo、zno、cr2o3、al2o3和适量助剂(v、mo、mn、mg、ce),具有较高的co转化率和较高的c
2+
醇选择性。但是,以上通过co加氢一步制备的混合醇碳数比较低(一般为c1~c6),基本上得不到高附加值的c
6+
高碳醇,附加值低的甲醇比例较高(>40%),严重制约了该技术的经济性。
7.中国科学院大连化学物理研究所开发了一系列活性炭负载co基催化剂用于合成气一步法直接合成直链混合伯醇。其中c2~c5低碳混合醇和c
6+
高碳混合醇都是重要的高附加值精细化学品。该催化体系所需的反应条件温和(220℃,3.0mpa,1000~4000h-1
,h2/co=2/1)。该过程的工艺简单、设备投资少、产品附加值高,费托合成产物中烃类(主要是石脑油和柴油)和混合伯醇(c1~c
18
oh)的质量比约为1:1,而且甲醇的选择性低。专利us 7670985、us 7468396和cn101310856公开了其催化剂体系为活性炭负载的co基催化剂,在其催化作用下co加氢可以直接合成高碳混合伯醇,液体产品中c2~c
18
醇的选择性高达60%,其中甲醇在醇中的分布只占约2~4%。该团队从催化剂组成和结构出发,对催化反应机理都进行了深入的研究,发现助剂的种类和含量对反应性能的影响十分显著。该催化体系可以由合成气直接制备高碳醇的原因是在反应过程中原位形成co2c物种,co2c物种可以非解离吸附co,促进co插入;而金属co上进行co解离吸附,促进碳链增长。co-co2c界面结构构成了高碳醇形成的双活性中心。
技术实现要素:
8.本发明的目的是,针对现有技术存在的不足,开发一种同时提高费托合成产物中烯烃和含氧化合物选择性的催化剂,催化剂活性提高接近一倍,烯烃和含氧化合物的总选择性达到60%以上(以碳数计),液相产物中烯烃和含氧化合物所占比例不低于70wt%(以质量计),以期从合成气出发,一步实现油-醇联产。
9.为实现上述目标,本发明所采用的技术方案具体包括:
10.一种合成气油-醇联产的催化剂,表示为co-x-y-z/ac,其特征在于,催化剂活性组分为金属co和反应过程中生成的co2c中的一种或二种以上,第一助剂为x为zr、zn、cu、cr中的一种或二种以上,第二助剂y为mn、la、ce、mo、v中的一种或二种以上,第三助剂z为li、na、k、mg、ca、sr中的一种或二种以上。催化剂中金属co的含量为催化剂重量的5~30wt%(优选10~20wt%),第一助剂x的含量为催化剂重量的0.5~10wt%(优选0.5~5wt%),第二助剂y的含量为催化剂重量的0~5wt%(优选0.5~3wt%),第三助剂z的含量为催化剂重量的0~3wt%(优选0.1~2wt%)。载体为杏核活性炭和椰壳活性炭中的一种或二种以上,该活性炭的粒径为20~400目,比表面积为200~1500m2/g,平均孔径为0.1~20nm,总孔容为0.1~2.0ml/g。
11.上述催化剂制备方法包括以下步骤:
12.(1)活性炭原料先在质量分数为5%~15%的盐酸或硝酸溶液中煮沸洗涤处理1小时以上,再用去离子水煮沸洗涤处理1小时以上,然后在110~130℃的空气气氛中烘干12~24小时,粉碎至颗粒20~400目,备用;
13.(2)用活性组分的可溶性盐中的一种或二种以上和助剂x的可溶性盐中的一种或二种以上的混合水溶液,或用活性组分的可溶性盐中的一种或二种以上、助剂x的可溶性盐中的一种或二种以上和助剂y的可溶性盐中的一种或二种以上的混合水溶液,浸渍上述处理后的活性炭载体;浸渍后的催化剂前驱体先在室温下阴干6~12小时,然后在50~120℃的空气气氛中烘干8~48小时,制得半干基催化剂前驱体a;
14.(3)半干基催化剂前驱体a在氮气、氩气或氦气中的一种或二种气氛下,温度200~500℃(优选300~450℃),压力为常压,气体空速100~3000h-1(优选500~2500h-1),焙烧5
~25小时,得到干基催化剂b;
15.(4)然后用助剂y的可溶性盐中的一种或二种以上和助剂z的可溶性盐中的一种或二种以上的混合水溶液、或用助剂z的可溶性盐中的一种或二种以上的水溶液,浸渍上述处理后的干基催化剂b;浸渍后的催化剂前驱体先在室温下阴干6~12小时,然后在50~120℃的空气气氛中烘干8~48小时,制得半干基催化剂前驱体c;
16.(5)半干基催化剂前驱体c在氮气、氩气或氦气中一种或二种以上气氛下,温度200~500℃(优选250~430℃),压力为常压,气体空速100~3000h-1(优选300~2500h-1),焙烧5~25小时,得到干基催化剂d;
17.(6)干基催化剂d在含氢气氛中还原活化,含氢气氛为氢气或含氢混合气,含氢气氛中氢气体积含量为5~100%(优选10~95%),含氢混合气中的除氢气以外的气体为氮气、氩气或氦气中一种或二种以上,还原活化温度200~600℃(优选330~450℃),压力0.1~3.0mpa(优选0.1~1.0mpa),气体空速300~8000h-1(优选1000~3000h-1),还原活化4~96小时,制备得活化催化剂e;
18.(7)活化催化剂e还需经h2和co的混合气进行预处理;所述混合气中h2与co摩尔比为0.5~5.0(优选1.0~3.0),预处理温度为180~260℃(优选190~230℃),压力0.1~10.0mpa(优选1.0~6.0mpa),气体空速100~10000h-1(优选1000~5000h-1),处理时间2~120小时。
19.上述催化剂既可以应用于固定床反应器,又可以应用于浆态床反应器。应用于固定床反应器时,上述(5)、(6)、(7)步骤在固定床反应器中原位进行;应用于浆态床反应器时,(5)、(6)步骤在流化床反应器中进行,完成后将催化剂转移到浆态床反应器中进行(7)步骤。催化剂转移时应采用惰性气体保护输送。
20.合成气油-醇联产的反应器形式采用固定床反应器或浆态床反应器,反应条件为温度180~250℃(优选190~230℃),压力0.1~5.0mpa(优选1.0~4.0mpa),空速500~6000h-1
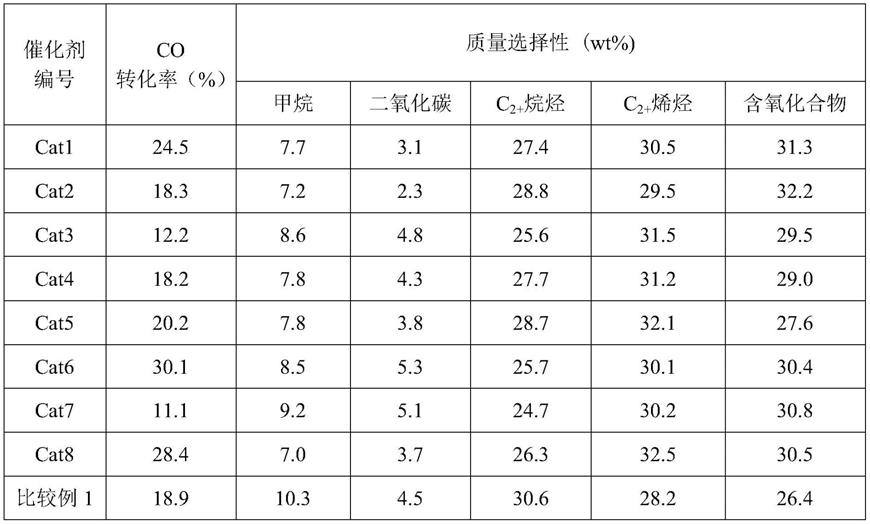
(优选500~3500h-1
),h2/co摩尔比0.5~5.0(优选1.0~3.0)。原料合成气(h2和co的混合气)的进料方式采取连续进料,其在催化剂床层连续进行co加氢反应,反应生成的气体产物和液体产物连续出料,采用热罐收集高沸点的重质组分,采用冷罐收集低沸点的轻质组分,热罐加热温度维持在100~130℃,冷罐温度维持在0~10℃。
21.费托合成产物中清洁液体燃料为碳数为5个碳原子以上的直链烷烃,可作为调和汽柴油组分;碳数2~4的直链烷烃可作为液化石油气组分;烯烃为碳数2~20的直链烯烃,含氧化合物为碳数2~20的直链混合伯醇、醛等;产物中烯烃和混合醇所占比例不低于60%(以碳数计);液相产物中烯烃和混合醇所占比例不低于70wt%(以质量计)。
22.本发明与现有技术相比具有以下优点:
23.①
本发明提供的催化剂,可以同时提高合成产物中烯烃和含氧化合物的选择性,其总质量在液体产物中所占比例不低于70wt%。同时产物中含有一定比例的直链烷烃,可以实现以合成气为原料的油-醇联产过程。
24.②
本发明提供的催化剂,利用co2c物种的类贵金属性质,实现co非解离吸附和插入碳链的作用,不使用贵金属,催化剂价格降低,有利于大批量工业化生产。
25.③
本发明提供的催化剂,载体为活性炭,在催化剂活化和反应过程中,不与金属co反应生成硅酸钴、铝酸钴等难还原物质,有利于提高co的分散度和还原度,提高催化剂寿
命。同时,以碳材料为催化剂载体易于回收其中的金属钴,实现催化剂中金属的循环利用,提高了催化剂的经济性。
26.本发明与中科院大连化学物理研究所前期公布的活性炭负载钴基费托合成催化剂相比,其优势在于,在保持了其较高的烯烃和含氧化合物选择性基础上,大大提高了催化剂的活性和稳定性,产生的烯烃可以进一步通过氢甲酰化转化成醇、醛等高值化学品,同时抑制了ch4的形成,提高了co加氢产物的综合价值,实现以合成气为原料的油-醇联产过程。
具体实施方式
27.以下结合实施例和附表对本发明作进一步的说明,并不对本发明进行任何限制。
28.实施例1~6选用粒径为120~200目,比表面积960m2/g,平均孔径3.8nm,孔容0.6ml/g的椰壳活性炭为载体,将其在90℃去离子水中反复洗涤20次,在393k下干燥20小时,得到纯化后的活性炭载体。
29.实施例1
30.制备10co2zn0.5mn0.2na/ac催化剂(元素前数值表示其于催化剂中的质量百分含量),标记为cat1。
31.将4.94g六水合硝酸钴、0.91g六水合硝酸锌、0.23g四水乙酸锰溶于10g水中,制成浸渍液。用该浸渍液在室温298k下浸渍8.73g活性炭载体后阴干约10小时,至催化剂表面没有可流动水分后,置于313k烘箱中烘干24h,取出后在氩气中,逐渐升温至573k焙烧20小时得到干基催化剂前驱体a。再将0.074g硝酸钠溶于10g水中,制成浸渍液。用该浸渍液在室温298k下浸渍干基催化剂前驱体a后阴干约10小时,至催化剂表面没有可流动水分后,置于313k烘箱中烘干24h,取出后在氩气中,逐渐升温至573k焙烧20小时得到干基催化剂前驱体b。
32.将干基催化剂前驱体b取出装入直径为9mm的固定床反应器中,填充量为2ml,空速2000h-1
。催化剂在氢气中逐渐升温至673k还原15小时,降温至423k,切换合成气(h2/co=2/1),压力2mpa,升温至493k进行ft反应。稳定12小时后,每隔24小时取样分析尾气中各组分浓度、液相产物组成以及液相产物收率。所得结果列于附表1。
33.实施例2
34.制备10co3cu1ce0.3k/ac催化剂,标记为cat2。
35.将4.94g六水合硝酸钴、1.14g三水合硝酸铜和0.31g六水合硝酸铈溶于10g水中,制成浸渍液。用该浸渍液在室温298k下浸渍8.57g活性炭载体,阴干约10小时,至催化剂表面没有可流动水分后,置于313k烘箱中烘干24小时,取出后在氮气中,逐渐升温至573k焙烧20小时,得到干基催化剂前驱体a。再将0.078g硝酸钾溶于10g水中,制成浸渍液。用该浸渍液在室温298k下浸渍干基催化剂前驱体a后阴干约10小时,至催化剂表面没有可流动水分后,置于313k烘箱中烘干24h,取出后在氩气中,逐渐升温至573k焙烧20小时得到干基催化剂前驱体b。
36.将干基催化剂前驱体b取出装入直径为9mm的固定床反应器中,填充量为2ml,空速2000h-1
。催化剂在氢气中逐渐升温至673k还原20小时,降温至423k,切换合成气(h2/co=2/1),压力2mpa,升温至493k进行ft反应。稳定12小时后,每隔24小时取样分析尾气中各组分浓度、液相产物组成以及液相产物收率。所得结果列于附表1。
37.实施例3
38.制备10co2zr1la1li/ac催化剂,标记为cat3。
39.将4.94g六水合硝酸钴和0.94g四水硝酸锆于10克水中,制成浸渍液。用该浸渍液在室温298k下浸渍8.40g活性炭载体后阴干约10小时,至催化剂表面没有可流动水分后,置于313k烘箱中烘干24小时,取出后在氮气中,逐渐升温至573k焙烧20小时,得到干基催化剂前驱体a。再将0.31g六水合硝酸镧和0.99g硝酸锂溶于10g水中,制成浸渍液。用该浸渍液在室温298k下浸渍干基催化剂前驱体a后阴干约10小时,至催化剂表面没有可流动水分后,置于313k烘箱中烘干24h,取出后在氩气中,逐渐升温至573k焙烧20小时得到干基催化剂前驱体b。
40.将干基催化剂前驱体b取出装入直径为9mm的固定床反应器中,填充量为2ml,空速2000h-1
。催化剂在氢气中逐渐升温至673k还原20小时,降温至423k,切换合成气(h2/co=2/1),压力2mpa,升温至493k进行ft反应。稳定12小时后,每隔24小时取样分析尾气中各组分浓度、液相产物组成以及液相产物收率。所得结果列于附表1。
41.实施例4
42.制备10co2cr0.5mo0.3mg/ac催化剂,标记为cat4。
43.将4.94g六水合硝酸钴和1.54g九水硝酸铬溶于10g水中,制成浸渍液。用该浸渍液在室温298k下浸渍8.72g活性炭载体后阴干约10小时,至催化剂表面没有可流动水分后,置于313k烘箱中烘干24小时,取出后在氮气中,逐渐升温至573k焙烧20小时,得到干基催化剂前驱体a。再将0.10g钼酸铵和0.18g硝酸镁溶于10g水中,制成浸渍液。用该浸渍液在室温298k下浸渍干基催化剂前驱体a后阴干约10小时,至催化剂表面没有可流动水分后,置于313k烘箱中烘干24h,取出后在氩气中,逐渐升温至573k焙烧20小时得到干基催化剂前驱体b。
44.将干基催化剂前驱体b取出装入直径为9mm的固定床反应器中,填充量为2ml,空速2000h-1
。催化剂在氢气中逐渐升温至673k还原20小时,降温至423k,切换合成气(h2/co=2/1),压力2mpa,升温至493k进行ft反应。稳定12小时后,每隔24小时取样分析尾气中各组分浓度、液相产物组成以及液相产物收率。所得结果列于附表1。
45.实施例5
46.制备10co4cu1v0.5ca/ac催化剂,标记为cat5。
47.将4.94g六水合硝酸钴和1.52g三水合硝酸铜溶于10克水中,制成浸渍液。用该浸渍液在室温298k下浸渍8.45g活性炭载体后阴干约10小时,至催化剂表面没有可流动水分后,置于313k烘箱中烘干24小时,取出后在氮气中,逐渐升温至573k焙烧20小时,得到干基催化剂前驱体a。再将0.23g偏钒酸铵和0.21g硝酸钙溶于10g水中,制成浸渍液。用该浸渍液在室温298k下浸渍干基催化剂前驱体a后阴干约10小时,至催化剂表面没有可流动水分后,置于313k烘箱中烘干24h,取出后在氩气中,逐渐升温至573k焙烧20小时得到干基催化剂前驱体b。
48.将干基催化剂前驱体b取出装入直径为9mm的固定床反应器中,填充量为2ml,空速2000h-1
。催化剂在氢气中逐渐升温至673k还原20小时,降温至423k,切换合成气(h2/co=2/1),压力2mpa,升温至493k进行ft反应。稳定12小时后,每隔24小时取样分析尾气中各组分浓度、液相产物组成以及液相产物收率。所得结果列于附表1。
49.实施例6
50.制备10co3zn1mn0.5li/ac催化剂,标记为cat6。
51.将4.94g六水合硝酸钴和1.36g六水合硝酸锌溶于10g水中,制成浸渍液。用该浸渍液在室温298k下浸渍8.59g活性炭载体后阴干约10小时,至催化剂表面没有可流动水分后,置于313k烘箱中烘干24小时,取出后在氮气中,逐渐升温至573k焙烧20小时,得到干基催化剂前驱体a。再将0.45g乙酸锰和0.50g硝酸锂溶于10g水中,制成浸渍液。用该浸渍液在室温298k下浸渍干基催化剂前驱体a后阴干约10小时,至催化剂表面没有可流动水分后,置于313k烘箱中烘干24h,取出后在氩气中,逐渐升温至573k焙烧20小时得到干基催化剂前驱体b。
52.将干基催化剂前驱体b取出装入直径为9mm的固定床反应器中,填充量为2ml,空速2000h-1
。催化剂在氢气中逐渐升温至673k还原20小时,降温至423k,切换合成气(h2/co=2/1),压力2mpa,升温至493k进行ft反应。稳定12小时后,每隔24小时取样分析尾气中各组分浓度、液相产物组成以及液相产物收率。所得结果列于附表1。
53.实施例7~8
54.选用杏核活性炭作为载体,载体比表面积912m2/g,平均孔径3.5nm,孔容0.43ml/g,将其在煮沸的去离子水中反复洗涤20次,在393k下干燥20小时,得到纯化后的活性炭载体。
55.按照与实施例3相似的方法制备和评价10co2zr1la1li/ac催化剂,标记为cat7,所得结果列于附表1。
56.按照与实施例6相似的方法制备和评价10co2zn1mn0.1li/ac催化剂,标记为cat8,所得结果列于附表1。
57.比较例1
58.改用专利cn201210292413提供的配方和方法,制备催化剂,采用相似的方法对其进行反应评价,并与实施例进行比较。所得结果列于附表1。
59.比较例1:10co0.1al/ac
60.附表1
61.co-x-y-z/ac催化剂上合成气油-醇联产催化剂性能
[0062][0063]
从实施例和比较例的结果看,本专利提供的催化剂在保持了其较高的烯烃和含氧化合物选择性基础上,提高了催化剂的活性和稳定性,产生的烯烃可以进一步通过氢甲酰化转化成醇、醛等高值化学品,同时抑制了ch4的形成,提高了co加氢产物的综合价值,实现以合成气为原料的油-醇联产过程。而与其他以低碳烯烃为主产物的催化剂相比,本专利提供的催化剂能够同时产生大量含氧化合物,具有更高的原子经济性和附加价值。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1