核苷类亲水作用色谱固定相及制备方法及应用与流程
本发明涉及色谱固定相,具体地说是核苷类亲水作用色谱固定相及制备方法及应用。
背景技术:
近年来,随着蛋白质组学、生物医学和环境监测等研究领域的快速发展,强极性和亲水性小分子物质迅速成为分析化学和生物化学领域的重要研究对象,准确、高效的分离这些物质就变得极其重要。这些物质通常很难在反相液相色谱柱上得到有效分离,正相液相色谱往往使用一些昂贵且危险的流动相,如正己烷或环己烷来分离这些极性物质。然而,在这种非水溶性的流动相中,许多极性和亲水性化合物往往难以溶解,限制了正相色谱的应用。因此,亲水作用色谱(hydrophilicinteractionchromatography,hilic)被视为分离强极性和亲水性化合物的一种替代方案。与流动相为非极性有机相的正相色谱不同,hilic的流动相往往由水或缓冲液和60-95%的乙腈或甲醇组成,其类似于反相色谱的流动相。强极性分析物在此类流动相中有较大的溶解度,因此,hilic已成为一种非常适合于极性和亲水性物质分离及分析的液相色谱技术。目前,已有众多亲水作用色谱固定相被商业化。根据其键合基团的结构可分为:醇型、胺型、两性离子型、氰基型、多糖型和纯硅胶型等。由于实际样品的复杂性和分离要求的多样性,目前还没有一种亲水作用色谱能够满足大部分极性化合物分离的要求,因此开发新型亲水作用色谱固定相是一项十分重要的工作。
腺苷、胞苷、鸟苷和尿苷是一类遍布人体细胞的内源性核苷,由嘌呤的n-9或嘧啶的n-1与d-核糖的c-1通过β糖苷键连接而成的化合物。这类化合物分子中含有一分子的碱基和一分子的糖环,它们自身带有的羟基和氨基使其具有良好的亲水性。多年来,核苷碱基类的物质多作为分析物用于hilic保留模式的研究,还未有将其制备成亲水作用色谱固定相的报道。根据核苷类化合物自身的性质,将它们制备成亲水作用色谱固定相并用于极性和亲水性物质的分离,是十分有意义的工作。
技术实现要素:
本发明的目的是提供一种核苷类亲水作用色谱固定相及制备方法及应用,核苷类化合物包括腺苷、胞苷、鸟苷和尿苷。
为实现上述目的,本发明采用技术方案为:
本发明公开了核苷类亲水作用色谱固定相,核苷类化合物包括腺苷、胞苷、鸟苷和尿苷,固定相为式一所示:
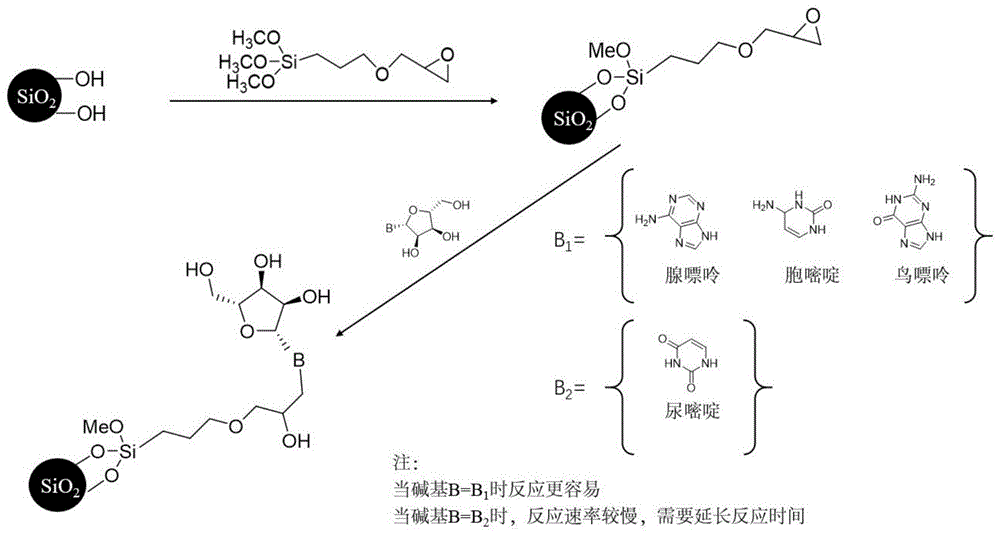
作为进一步地改进,本发明所述固定相为先将硅烷偶联剂键合到硅胶载体上制备硅烷化硅胶,而后利用硅烷偶联剂中的环氧烷和氨基的开环反应,将核苷键合到硅胶载体上,进而获得式一所示的核苷类亲水作用色谱固定相。
本发明还公开了一种制备核苷类亲水作用色谱固定相的制备方法:
1)将硅胶经硅烷化修饰制备硅烷化硅胶
将硅胶,有机溶剂,硅烷偶联剂按质量比为1:(7-30):(2.3-3)混合,混合后于90-130℃条件下反应12-48h,反应结束后过滤、洗涤,洗涤后真空干燥既得硅烷化硅胶;其中,有机溶剂为无水甲苯,每克硅胶所需的质量为5~30g,约10~40ml;反应完成后用丙酮抽提12-24h,洗涤后45~100℃下真空干燥12~24h。在保证硅胶可以均匀分散在有机溶剂的情况下,适当增加硅烷偶联剂的加入量,可以提高硅烷偶联剂键合到硅胶表面的效率。
2)硅烷化硅胶与核苷类化合物的反应
将上述获得的硅烷化硅胶与核苷混合,加入一定量的水,在60-85℃条件下反应30-90h,反应结束后离心,沉淀洗涤后真空干燥,既得亲水作用色谱固定相。
作为进一步地改进,本发明所述的步骤2)中,核苷类化合物加入的物质的量为硅烷化硅胶上环氧烷的物质的量的1-10倍,每克硅烷化硅胶所需水量为8-15ml。
作为进一步地改进,本发明所述步骤1)中有机溶剂为无水甲苯,每克硅胶所需要的质量为5-30g。
作为进一步地改进,本发明所述的步骤1)中所用的硅烷偶联剂为3-(2,3-环氧丙氧)丙基三甲氧基硅烷,其结构式为:
其反应式为:
作为进一步地改进,本发明所述的步骤1)中所用的硅胶为微球型,是为粒径和孔径均匀的全多孔硅胶球状体,其粒径为3~20um,孔径为
本发明还公开了四种核苷亲水作用色谱固定相的应用,所述式一所示化合物在作为亲水作用色谱中的固定相对极性和亲水性化合物分离中的应用。
作为进一步地改进,本发明所述的极性化合物为核苷碱基类化合物。其中,极性化合物为核苷与碱基如鸟苷、胞苷、腺苷、尿苷、尿嘧啶、腺嘌呤、胞嘧啶和胸腺嘧啶。
分离的条件为温度(t=25~50℃)、流动相:乙腈:水或buffer溶液体积比为70:30~95:5(其中buffer溶液包括:甲酸铵溶液、乙酸铵溶液、乙酸钠溶液、磷酸二氢钠溶液、磷酸氢二钠溶液)、流速:0.1~1.2ml/min、检测波长:254nm。
本发明具有以下优点:
本发明获得的核苷类键合硅胶色谱固定相应用在亲水作用色谱中,能够较好的分离极性化合物;具体为:
1)本发明的固定相原料易得,合成步骤简单。
2)本发明的固定相使用四种核苷(腺苷、胞苷、鸟苷和尿苷)作为极性功能基团,其结构简单,稳定性好。
3)本发明的固定相以高比例的乙腈流动相进行分离,避免了使用正相色谱需要用到的强毒性和高危性有机试剂,适用于核苷碱基类物质的分离分析。
4)本发明的固定相采用环氧烷与氨基开环反应作为键合方式,可以在温和的条件下实现高选择性和高转化率的固载。
附图说明
图1为本发明核苷类亲水作用色谱固定相的合成路线图;
图2为本发明实施例1和2提供的固定相红外表征图;
图3为本发明实施例1和2提供的固定相亲水色谱模式图;
图4为本发明实施例1提供的固定相在hilic模式下分离八种核苷碱基的色谱分离图。
具体实施方式
发明涉及色谱固定相,具体说是四种核苷(腺苷、胞苷、鸟苷和尿苷)亲水作用色谱固定相及其制备和应用。固定相为式一所示,通过3-(2,3-环氧丙氧)丙基三甲氧基硅烷对硅胶表面进行改性,利用环氧烷与氨基的开环反应将四种核苷分别键合到改性硅胶表面,得到预期固定相。本发明固定相原料易得,操作步骤简单,稳定性好,具有良好的应用前景。
以下结合附图说明给出本发明的实施例,对本发明做进一步的说明;提供的实施例仅限于说明本发明,而非对本发明实施范围的限定。
本发明涉及四种亲水作用色谱固定相,通过3-(2,3-环氧丙氧)丙基三甲氧基硅烷对硅胶表面进行改性,利用环氧烷与氨基的开环反应将四种核苷分别键合到改性硅胶表面,得到预期固定相。本发明固定相原料易得,操作步骤简单,应用广泛,稳定性好。所述的核苷类亲水作用色谱固定相在亲水作用色谱模式下可有效的用于对极性化合物(核苷碱基类物质)的分离。
图1为本发明四种核苷亲水作用色谱固定相的合成路线图,第一步,将3-(2,3-环氧丙氧)丙基三甲氧基硅烷键合到硅胶表面,制备3-(2,3-环氧丙氧)丙基三甲氧基硅烷烷基化硅胶;第二步,通过环氧烷和氨基的开环反应,分别将四种核苷键合到硅烷化硅胶表面制备核苷类固定相。
实施例1
腺苷亲水作用色谱固定相的制备
(1)3-(2,3-环氧丙氧)丙基三甲氧基硅烷烷基化硅胶的制备
在100ml三口烧瓶中依次加入4.5g活化的粒径为5um的球形硅胶颗粒、50ml无水甲苯、17.85mmol(约4.58ml)3-(2,3-环氧丙氧)丙基三甲氧基硅烷,在110℃、氩气保护的条件下反应24小时。反应完毕后,用丙酮抽提12-24h,洗涤后45~100℃下真空干燥12~24h,即得3-(2,3-环氧丙氧)丙基三甲氧基硅烷烷基化硅胶;
(2)3-(2,3-环氧丙氧)丙基三甲氧基硅烷烷基化硅胶键合腺苷的制备
取4.5g干燥后的3-(2,3-环氧丙氧)丙基三甲氧基硅烷烷基化硅胶置于100ml三口烧瓶中,加入50ml水、2.4g腺苷,在65℃、氩气保护条件下反应60h,反应结束后,依次用1200ml水、150ml甲醇、150ml丙酮洗涤,室温下真空干燥24h,即得腺苷固定相,其结构为:
图2a为本发明实施例1提供的固定相红外表征图,由sio2的红外光谱图可以发现,在1100cm-1处的宽带吸收峰是si-o-si的吸收峰,在993cm-1和3466cm-1处出现了硅羟基的吸收峰。与之相比,它们在sio2-gptms,gptms是3-(2,3-环氧丙氧)丙基三甲氧基硅烷的英文简写,谱图中吸收强度明显减少,表明gptms已键合到sio2的表面。由于-oh和-nh-基团的伸缩振动,在ttfhps1谱图中3466cm-1处的吸收峰的强度再次增强,表明腺苷键合到sio2-gptms的表面。
实施例2
胞苷亲水作用色谱固定相的制备
(1)3-(2,3-环氧丙氧)丙基三甲氧基硅烷烷基化硅胶的制备
在100ml三口烧瓶中依次加入4.5g活化的粒径为5um的球形硅胶颗粒、50ml无水甲苯、17.85mmol(约4.58ml)3-(2,3-环氧丙氧)丙基三甲氧基硅烷,在110℃、氩气保护的条件下反应24h。反应完毕后,用丙酮抽提12-24h,洗涤后45~100℃下真空干燥12~24h,即得3-(2,3-环氧丙氧)丙基三甲氧基硅烷烷基化硅胶;
(2)3-(2,3-环氧丙氧)丙基三甲氧基硅烷烷基化硅胶键合胞苷的制备
取4.5g干燥后的3-(2,3-环氧丙氧)丙基三甲氧基硅烷烷基化硅胶置于100ml三口烧瓶中,加入50ml水、2.2g胞苷,在65℃、氩气保护条件下反应60h,反应结束后,依次用1200ml水、150ml甲醇、150ml丙酮洗涤,室温下真空干燥24h,即得胞苷固定相,其结构为:
图2b为本发明实施例2提供的固定相红外表征图,由sio2的红外光谱图可以发现,在1100cm-1处的宽带吸收峰是si-o-si的吸收峰,在993cm-1和3463cm-1处出现了硅羟基的吸收峰。与之相比,它们在sio2-gptms,gptms是3-(2,3-环氧丙氧)丙基三甲氧基硅烷的英文简写,谱图中吸收强度明显减少,表明gptms已键合到sio2的表面。由于-oh和-nh-基团的伸缩振动,在ttfhps2谱图中3463cm-1处的吸收峰的强度再次增强,表明胞苷键合到sio2-gptms的表面。
实施例3
鸟苷亲水作用色谱固定相的制备
(1)3-(2,3-环氧丙氧)丙基三甲氧基硅烷烷基化硅胶的制备
在100ml三口烧瓶中依次加入4.5g活化的粒径为5um的球形硅胶颗粒、50ml无水甲苯、17.85mmol(约4.58ml)3-(2,3-环氧丙氧)丙基三甲氧基硅烷,在110℃、氩气保护的条件下反应24h。反应完毕后,用丙酮抽提12-24h,洗涤后45~100℃下真空干燥12~24h,即得3-(2,3-环氧丙氧)丙基三甲氧基硅烷烷基化硅胶;
(2)3-(2,3-环氧丙氧)丙基三甲氧基硅烷烷基化硅胶键合鸟苷的制备
取4.5g干燥后的3-(2,3-环氧丙氧)丙基三甲氧基硅烷烷基化硅胶置于100ml三口烧瓶中,加入50ml水、2.5g鸟苷,在65℃、氩气保护条件下反应60h,反应结束后,依次用1200ml水、150ml甲醇、150ml丙酮洗涤,室温下真空干燥24h,即得鸟苷固定相,其结构为:
实施例4
尿苷亲水作用色谱固定相的制备
(1)3-(2,3-环氧丙氧)丙基三甲氧基硅烷烷基化硅胶的制备
在100ml三口烧瓶中依次加入4.5g活化的粒径为5um的球形硅胶颗粒、50ml无水甲苯、17.85mmol(约4.58ml)3-(2,3-环氧丙氧)丙基三甲氧基硅烷,在110℃、氩气保护的条件下反应24h。反应完毕后,用丙酮抽提12-24h,洗涤后45~100℃下真空干燥12~24h,即得3-(2,3-环氧丙氧)丙基三甲氧基硅烷烷基化硅胶;
(2)3-(2,3-环氧丙氧)丙基三甲氧基硅烷烷基化硅胶键合尿苷的制备
取4.5g干燥后的3-(2,3-环氧丙氧)丙基三甲氧基硅烷烷基化硅胶置于100ml三口烧瓶中,加入50ml水、2.2g尿苷,在65℃、氩气保护条件下反应90h,反应结束后,依次用1200ml水、150ml甲醇、150ml丙酮洗涤,室温下真空干燥24h,即得尿苷固定相,其结构为:
应用例1
下述应用例以实施例1或2获得的固定相为例,对本发明的固定相进行相应的效果评价,具体为:
把实施例1所得的腺苷固定相填料填装于4.6mm*250mm,i.d.的不锈钢色谱柱中,制得的色谱柱用于测试其在亲水色谱模式下的亲水色谱保留行为。
以8种核苷碱基(尿嘧啶、尿苷、腺苷、腺嘌呤、胞嘧啶、胞苷、鸟苷、胸腺嘧啶)作为分析物,流动相为两相,分别为乙腈,水。流动相条件分别为乙腈/水(65%/35%),乙腈/水(70%/30%,v/v),乙腈/水(75%/25%,v/v),乙腈/水(80%/20%,v/v),乙腈/水(85%/15%,v/v),乙腈/水(90%/10%,v/v),流速1.0ml/min,柱温为30℃,检测波长为254nm。亲水色谱保留图如图3a所示,由图可知,随着流动相中水体积分数的减少,保留时间增加,这是典型的亲水色谱保留特征,由此可以说明腺苷固定相具有典型的亲水作用色谱保留行为。
应用例2
把实施例2所得的胞苷固定相填料填装于4.6mm*250mm,i.d.的不锈钢色谱柱中,制得的色谱柱用于测试其在亲水色谱模式下的亲水色谱保留行为。
以8种核苷碱基(尿嘧啶、尿苷、腺苷、腺嘌呤、胞嘧啶、胞苷、鸟苷、胸腺嘧啶)作为分析物,流动相为两相,分别为乙腈,水。流动相条件分别为乙腈/水(70%/30%,v/v),乙腈/水(75%/25%,v/v),乙腈/水(80%/20%,v/v),乙腈/水(85%/15%,v/v),乙腈/水(90%/10%,v/v),流速1.0ml/min,柱温为30℃,检测波长为254nm。亲水色谱保留图如图3b所示,由图可知,随着流动相中水体积分数的减少,保留时间增加,这是典型的亲水色谱保留特征,由此可以说明胞苷固定相具有典型的亲水作用色谱保留行为。
应用例3
使用实施例1所制备的色谱柱在亲水作用色谱模式下分离核苷碱基类物质。流动相为两相,分别为乙腈,水。流动相条件为乙腈和水的体积比为90/10,流速1.0ml/min,柱温为30℃,检测波长为254nm。色谱图如图4所示(1.胸腺嘧啶2.尿嘧啶3.尿苷4.腺苷5.腺嘌呤6.胞嘧啶7.胞苷8.鸟苷)。分离结果表明,在亲水模式下,8种物质在本发明所提供的腺苷柱上可以实现基线分离,表现出典型的亲水作用模式特征和良好的分离选择性。
最后,还需要注意的是,以上列举的仅是本发明的具体实施例子。显然,本发明不限于以上实施例子,还可以有许多变形。本领域的普通技术人员能从本发明公开的内容直接导出或联想到的所有变形,均应认为是本发明的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!