一种多相催化剂的制备方法及其应用与流程
[0001]
本发明涉及催化剂技术领域,尤其是一种多相催化剂的制备方法及其应用。
背景技术:
[0002]
随着石油化工、农药、医药等产业的高速发展,不同种类的生产废水和生活污水尚未达标,便直接排入了地表水体,导致了严重的水污染,加剧了水资源短缺的矛盾,严重影响到居民饮水安全和身体健康。在现有高级氧化水处理技术中,均相芬顿反应(fe
2+
+h2o2)因其反应中fe
2+
能迅速活化h2o2,产生强氧化性的
·
oh而广泛应用于水中难降解有机污染物的降解,以提高废水可生化性。然而,在实际工程中,该技术因反应条件苛刻,ph适用范围窄(2~3)、反应后产生铁泥等缺陷而面临诸多限制。因此,有必要进行多相芬顿催化剂的研发。经研究发现,多相芬顿反应不仅具有高效的污染物降解能力,而且能克服经典的均相芬顿反应所存在的诸多瓶颈,实现活性组分固载化,使催化剂易于回收,可大大降低水处理的成本。
[0003]
然而,多相芬顿催化剂目前仍存在催化剂稳定性较低以及h2o2利用率较低等问题。有学者发现,通过“金属-载体”间的原位掺杂可使活性组分与载体发生络合,提高催化剂的稳定性。但在传统的络合方法中,催化剂的活性组分容易包裹在催化剂内部结构中,无法充分与h2o2和污染物接触。此外,在反应过程中过量的h2o2会被产生
·
oh氧化,产生超氧自由基(
·
o
2-),并且
·
o
2-在体系中最终被分解为o2,导致其有效利用率低,造成浪费。
技术实现要素:
[0004]
基于此,本发明的目的在于克服上述现有技术的不足之处而提供一种使用聚多巴胺修饰氮化碳载体络合活性组分(fe),同时,操作简便、成本低,制得的催化剂催化性能稳定的多相催化剂的制备方法。
[0005]
为实现上述目的,本发明所采取的技术方案为:一种多相催化剂的制备方法,包括如下步骤:
[0006]
(1)制备氮化碳纳米片:将尿素焙烧,得到氮化碳纳米片;
[0007]
(2)将盐酸多巴胺、步骤(1)中制备得到的氮化碳纳米片和铁源依次加入磷酸缓冲液中,搅拌溶解,超声,得到混合液a;
[0008]
(3)将步骤(2)中制备得到的混合液a在20-25℃下搅拌5h以上,得到固体产物b;
[0009]
(4)将步骤(3)中制备得到的固体产物b,洗涤、干燥,得到固体产物c;
[0010]
(5)将步骤(4)中制备得到的固体产物c在惰性气体下焙烧,降温后得到所述多相催化剂fe-gp@cn。
[0011]
本发明提供了一种fe表面修饰碳氮纳米片催化剂的合成方法。用尿素制备氮化碳、盐酸多巴胺(pda)作为碳源,fecl3作为铁源,通过自聚合及高温焙烧过程合成目标催化剂。
[0012]
本发明首次使用聚多巴胺修饰氮化碳载体络合活性组分(fe),从而使得活性组分
充分地暴露在载体材料表面,很好地解决了活性位点被包裹的问题。并且该复合材料,用于水处理中表现出优异的稳定性和催化性能,可快速活化水中溶解氧(do)分子,产生具有氧化性的超氧自由基(
·
o
2-),降解水中有机污染物。该体系有效利用do,取代了h2o2,降低了水处理成本,实现了芬顿反应理论上的突破。
[0013]
优选地,所述步骤(1)中,焙烧过程中的升温的速率为2.5-10℃/min,升温至500-600℃后,开始焙烧,焙烧的时间为2-4h。
[0014]
进一步优选地,所述步骤(1)中,焙烧过程中的升温的速率为2.5℃/min,升温至550℃后,开始焙烧,焙烧的时间为4h。
[0015]
优选地,所述步骤(2)中,盐酸多巴胺、氮化碳纳米片和铁源的重量比为:盐酸多巴胺:氮化碳纳米片:铁源=1:(1-3):(0.1-0.5);所述铁源为fecl3、fe(no3)3、fe2(so4)3、fepo4中的至少一种;超声的时间为5-10min。
[0016]
进一步优选地,所述铁源为fecl3;超声的时间为10min。
[0017]
优选地,所述步骤(2)中磷酸缓冲液的浓度为0.1-0.5mol/l,磷酸缓冲液的ph值=7.5-8.5。
[0018]
进一步优选地,所述步骤(3)中适宜的温度为25℃,搅拌的时间为10h。
[0019]
优选地,所述步骤(4)中,洗涤的次数为3-6次,干燥的温度为60-100℃。
[0020]
优选地,所述步骤(4)中,在真空下进行干燥。
[0021]
进一步优选地,所述步骤(4)中,洗涤的次数为6次,用超纯水和乙醇交替洗涤3次,干燥的温度为60℃。
[0022]
优选地,所述步骤(5)中,焙烧过程中的升温的速率为5-10℃/min,升温至700-900℃后,开始焙烧,焙烧的时间为0.5-1h。
[0023]
进一步优选地,所述步骤(5)中,焙烧的温度为900℃。
[0024]
同时,本发明还提供了一种上述的多相催化剂的制备方法制备得到的多相催化剂fe-gp@cn。
[0025]
本发明制备方法制得的fe表面修饰碳氮纳米片多相催化剂,具有丰富的孔隙结构;其表面fe物种修饰使该催化剂具有很好的磁性,易于收集;fe物种暴露在碳氮纳米片表面易于反应中与污染物及do分子的充分接触;在自组装过程中pda包在氮化碳表面,且fe物种(fe
2+
/fe
3+
)与pda发生络合,再经过高温焙烧使得pda碳化,从而形成稳定的fe-o-c键,使得该催化剂具有优异的稳定性;fe-o-c键以及fe0的形成使得催化剂表面形成极性中心,使得该催化剂可快速活化水中do分子形成氧化性的
·
o
2-,用于水体中有机污染物的氧化分解。
[0026]
进一步的,本发明还提供了所述的多相催化剂在构建连续流化床反应器中的应用。
[0027]
本发明利用上述催化剂构建流化床反应器,研究发现反应器连续运行仍可对污染物保持稳定的去除效果。本发明操作简便,成本低,难降解有机污染物去除效果明显,反应器催化性能稳定。利用该催化剂构建连续流流化床反应器,经实验发现,该反应器在长期实验中对污染物保持着稳定的去除效果。
[0028]
本发明提供的多相催化剂在构建连续流化床反应器中,通过蠕动泵控制底端进水-上端出水的连续流模式。所述进水停留时间为20-30min,优选30min;进水流速保证催化剂不被冲走即可。
[0029]
进一步的,本发明还提供了所述的多相催化剂在水处理中的应用。
[0030]
相对于现有技术,本发明的有益效果为:
[0031]
(1)本发明的fe物种表面修饰碳氮纳米片多相催化剂在反应过程不需要将体系的酸碱度(ph值)调至2~3这种苛刻条件,在中性室温条件下对于水中难生物降解的有机污染物的降解就具有很好的去除效果。
[0032]
(2)本发明的催化剂在降解污染物的过程中能够活化水中do,无需投加h2o2等氧化剂,打破了经典芬顿反应的理论瓶颈。
[0033]
(3)本发明的催化剂表面具有较多活性位点,比表面积大,其活性组分极大暴露在催化剂表面,具有对污染物和do的易接触性,不受空间位阻效应和毛细现象的明显影响。
[0034]
(4)本发明的催化剂在反应过程中不会产生铁泥等固体异物,不需要异物清除装置。
[0035]
(5)本发明在去除有机污染物的过程中具有很好的稳定性。
[0036]
(6)本发明的催化剂属于磁性固体催化剂,便于与水分离,便于回收循环利用。
[0037]
(7)本发明提出的连续流反应器操作简单,去污效果稳定。
附图说明
[0038]
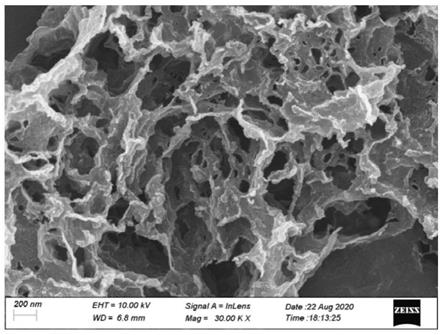
图1为实施例制得的fe-gp@cn的扫描电镜图;
[0039]
图2为实施例制得fe-gp@cn的x射线光电子能谱图;
[0040]
图3为实施例制得fe-gp@cn的电子顺磁共振谱图;
[0041]
图4为实施例制得fe-gp@cn与传统芬顿方法降解双酚a的对比图;
[0042]
图5为实施例制得fe-gp@cn对于不同污染物的降解曲线图;
[0043]
图6为实施例制得fe-gp@cn搭建的流化床图;
[0044]
图7为实施例制得fe-gp@cn流化床降解双酚a的柱形图。
具体实施方式
[0045]
为更好的说明本发明的目的、技术方案和优点,下面将结合附图和具体实施例对本发明作进一步说明。
[0046]
实施例1
[0047]
本发明多相催化剂的一种实施例,本实施例所述多相催化剂的制备方法,
[0048]
包括如下步骤:
[0049]
(1)制备氮化碳纳米片:将尿素焙烧,其中,焙烧的升温的速率为2.5℃/min,升温至500℃开始焙烧,焙烧的时间为2h,得到氮化碳纳米片;
[0050]
(2)将盐酸多巴胺、步骤(1)中制备得到的氮化碳纳米片和fe源依次加入磷酸缓冲液中,盐酸多巴胺、氮化碳纳米片和fe源的重量比为:盐酸多巴胺:氮化碳纳米片:fe(no3)3=1:3:0.1,搅拌溶解,超声,超声的时间为5min,得到混合液a;
[0051]
(3)将步骤(2)中制备得到的混合液a在25℃下搅拌5h,得到固体产物b;
[0052]
(4)将步骤(3)中制备得到的固体产物b,离心、洗涤干燥,洗涤的次数为3次,干燥的温度为60℃,得到固体产物c;
[0053]
(5)将步骤(4)中制备得到的固体产物c在惰性气体下焙烧,其中,焙烧的升温的速
率为5℃/min,升温至700℃开始焙烧,焙烧的时间为0.5h,降温后得到所述多相催化剂。
[0054]
将实施例1制备的fe-gp@cn-1催化剂,搭建反应器,通过蠕动泵控制底端进水-上端出水的连续流模式,其中进水停留时间为20min。
[0055]
实施例2
[0056]
本发明多相催化剂的一种实施例,本实施例所述多相催化剂的制备方法,包括如下步骤:
[0057]
(1)制备氮化碳纳米片:将尿素焙烧,其中,焙烧的升温的速率为2.5℃/min,升温至550℃开始焙烧,焙烧的时间为4h,得到氮化碳纳米片;
[0058]
(2)将盐酸多巴胺、步骤(1)中制备得到的氮化碳纳米片和fe源依次加入磷酸缓冲液中,盐酸多巴胺、氮化碳纳米片和fe源的重量比为:盐酸多巴胺:氮化碳纳米片:fecl3=1:1:0.1,搅拌溶解,超声,超声的时间为10min,得到混合液a;
[0059]
(3)将步骤(2)中制备得到的混合液a在25℃下搅拌10h,得到固体产物b;
[0060]
(4)将步骤(3)中制备得到的固体产物b,离心、洗涤干燥,用超纯水和乙醇交替洗涤3次,干燥的温度为60℃,得到固体产物c;
[0061]
(5)将步骤(4)中制备得到的固体产物c在惰性气体下焙烧,其中,焙烧的升温的速率为5℃/min,升温至900℃开始焙烧,焙烧的时间为1h,降温后得到所述多相催化剂。
[0062]
将实施例2制备的fe-gp@cn-2催化剂,搭建反应器,通过蠕动泵控制底端进水-上端出水的连续流模式,其中进水停留时间为30min。
[0063]
实施例3
[0064]
本发明多相催化剂的一种实施例,本实施例所述多相催化剂的制备方法,
[0065]
包括如下步骤:
[0066]
(1)制备氮化碳纳米片:将尿素焙烧,其中,焙烧的升温的速率为10℃/min,升温至600℃开始焙烧,焙烧的时间为4h,得到氮化碳纳米片;
[0067]
(2)将盐酸多巴胺、步骤(1)中制备得到的氮化碳纳米片和fe源依次加入磷酸缓冲液中,盐酸多巴胺、氮化碳纳米片和fe源的重量比为:盐酸多巴胺:氮化碳纳米片:fe2(so4)3=1:2:0.5,搅拌溶解,超声,超声的时间为10min,得到混合液a;
[0068]
(3)将步骤(2)中制备得到的混合液a在25℃下搅拌15h,得到固体产物b;
[0069]
(4)将步骤(3)中制备得到的固体产物b,离心、洗涤干燥,洗涤的次数为5次,干燥的温度为100℃,得到固体产物c;
[0070]
(5)将步骤(4)中制备得到的固体产物c在惰性气体下焙烧,其中,焙烧的升温的速率为10℃/min,升温至900℃开始焙烧,焙烧的时间为1h,降温后得到所述多相催化剂。
[0071]
将实施例3制备的fe-gp@cn-3催化剂,搭建反应器,通过蠕动泵控制底端进水-上端出水的连续流模式,其中进水停留时间为30min。
[0072]
实施例2制备的fe-gp@cn-2的分析检测如图1、图2、图3所示:
[0073]
图1为实施例2中制备得到的fe-gp@cn的sem图。从图中可以看出该催化剂具有层状、多孔结构。这主要是在高温焙烧过程中包覆在氮化碳纳米片表面的pda碳化而成,而且多孔的结构有利于在反应过程中污染物分子充分吸附在材料表面,并进一步被氧化分解。
[0074]
图2为实施例2中制备得到的fe-gp@cn的xps谱图。经过拟合分析得出fe-gp@cn表面的铁物种以fe0、fe(ii)、fe(iii)形式共存,多价态fe物种的形成使得催化剂表面自由电
子重新极化分布。
[0075]
如图3中fe-gp@cn的epr谱图所示,通过fe物种表面修饰,该催化剂表现出明显地自由电子信号,而没有经过fe修饰的载体材料则未检出自由电子信号。实施例1和实施例3制备得到的催化剂性能参数与实施例2相似,在此不一一赘述。
[0076]
对比例
[0077]
对比例为传统fenton氧化技术,投加fe盐。
[0078]
与对比例相比,本发明提供的体系不受溶液ph影响,不再需要将ph调至3~4,反应即可发生;其次,本发明提供的体系不需要氧化剂(h2o2)就能很好地对不同种类污染物进行氧化分解。突破了传统芬顿的诸多应用瓶颈,结果如图4所示。
[0079]
应用试验一
[0080]
将0.01g上述合成的催化剂投入50ml 10mg/l的污染物溶液中,维持自然ph值(约7.0),恒温35℃,在不投加氧化剂(h2o2)的情况下进行反应,分别在2,5,10,15,30分钟取样检测污染物的浓度,测试结果如图5和图7所示。
[0081]
应用实验二
[0082]
为考察该催化剂在长期使用中的稳定性,本发明中配置了bap 10mg/l的溶液,通过蠕动泵将其泵入反应器中,并在反应器中投加一定量的催化剂,使其浓度为0.2g/l,控制水力停留时间为30min。连续监测反应器出水的bpa浓度值。
[0083]
图5为实施例2制得fe-gp@cn对于不同污染物的降解曲线,如图所示,合成染料罗丹明b和亚甲基蓝在2分钟内就可完全脱色;内分泌干扰素双酚a在10分钟内完全去除;类抗生素环丙沙星在30分钟内可完全去除。由此得出,fe-gp@cn对水中难降解污染物有着优异的降解活性和较好的适应性。
[0084]
图7实施例2制得fe-gp@cn搭建流化床降解双酚a的柱形图,如图所示,在经过720个小时的连续运行,该催化剂仍能完全去除双酚a,说明该催化剂拥有很好的稳定性,为实际应用提供了理论基础。实施例1和实施例3制备得到的催化剂性能参数与实施例2相似,在此不一一赘述。
[0085]
最后所应当说明的是,以上实施例仅用以说明本发明的技术方案而非对本发明保护范围的限制,尽管参照较佳实施例对本发明作了详细说明,本领域的普通技术人员应当理解,可以对本发明的技术方案进行修改或者等同替换,而不脱离本发明技术方案的实质和范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1