一种CO-SCR脱硝催化剂及其制备方法和应用与流程
一种co-scr脱硝催化剂及其制备方法和应用
技术领域
1.本发明属于脱硝催化剂技术领域,具体涉及一种co-scr脱硝催化剂及其制备方法和应用。
背景技术:
2.氮氧化物(nox)作为主要大气污染物之一,是导致酸雨、二次细颗粒物、近地面臭氧形成和光化学烟雾的重要前体物。它不仅危害环境,还对工农业生产、人类健康存在严重威胁,其主要来源于火力发电厂、工业锅炉等排放的烟气和机动车排放的汽车尾气。近些年,随着人们环保意识的增强以及法律法规政策的日益严格,nox的脱除与减排工作已经提上环保部门以及各生产单位的日程。
3.目前,在众多脱硝技术中,国内外得到广泛应用的是以氨气选择性催化还原(nh
3-scr)技术为主,但其依然存在许多不足。nh
3-scr技术存在氨水存储以及易泄露等问题,因此研究人员一直在致力寻找可能的替代技术。co普遍存在于烧结、焦化等工业烟气中,因此利用co选择性催化还原nox(co-scr)被认为是极具应用潜力的可行技术之一,其显著优点是通过催化剂可实现co和no的同时去除。当前,研究较多的催化剂是贵金属(尤其是ir基催化剂),但由于其价格昂贵、高温失活、易中毒等问题难以在工业中实现应用。因此,开发经济高效的催化剂,尤其是金属氧化物催化剂,受到广泛关注。目前已开展研究的金属氧化物催化剂反应温度大多集中在200~350℃,且反应体系多数无o2存在。研究发现,在co-scr领域中,通常实际烟气工况中存在的氧气会降低催化剂的催化效率。在高温的有氧环境下,烟气中co易与o2发生反应生成co2,从而抑制nox的还原。因此,提高催化剂在氧存在下co对nox的还原性能,抑制还原剂co的氧化,对促进co-scr反应性能具有重要意义。
4.2co + o2→
2co2ꢀꢀꢀ
(1)2co + 2no
→
2co
2 + n2ꢀꢀ
(2)关于co还原no反应的反应机理,得到普遍认可的是no分子先裂解成n和o自由基,两个n自由基结合成吸附态n2分子,n自由基与吸附态no结合生成吸附态n2o分子,再次解析出n2分子。co还原no的反应可分为四个过程,分别为co、no分子的吸附、吸附态分子的解离、表面活性物质的重组和产物分子的脱附。在催化剂表面还原态的活性中心上no首先部分被还原为n2o,再进一步还原成n2。同时,催化剂表面还原态的活性中心被氧化,氧化后的催化剂活性中心再和吸附状态co反应,co被氧化成co2,再释放出还原态活性中心使得催化反应可以继续进行,反应过程如下。
5.第一步:co、no分子的吸附:co(g)
→
co(ads) (1)no(g)
→
no(ads) (2)第二步:吸附态分子的解离:no(ads)
→
n(ads) +o(ads) (3)第三、四步:表面活性物质的重组、产物分子的脱附:
2n(ads)
→
n2(g)
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
(4)n(ads)+no(ads)
→
n2o(g)
ꢀꢀꢀ
(5)co(ads) +o(ads)
→
co2(g)
ꢀꢀꢀ
(6)2no(ads)
→
n2(g)+2o(ads)
ꢀꢀꢀ
(7)2no(ads)
→
n2o(g)+o(ads)
ꢀꢀ
(8)中间产物n2o发生的反应:n2o(g)
→
n2o(ads)
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
(9)n2o(ads)
→
n2(g)+o(ads)
ꢀꢀꢀ
(10)o2的吸附能力强于no,它将与no竞争并吸附在催化剂上。这将导致吸附的no减少,吸附的o2增加。因此,o2存在将抑制co-scr的发生。另一方面o2会使金属氧化物上的氧空位减少甚至消失,导致co-scr反应活性降低。工业锅炉/窑炉烟气中往往含有一定浓度的o2,如何抑制o2对co-scr活性的影响具有重要意义。
技术实现要素:
6.针对现有技术的不足,本发明提供了一种co-scr脱硝催化剂及其制备方法和应用。本发明提供的脱硝催化剂在o2存在下具有良好的co-scr脱硝性能,避免了o2对co-scr过程的不利影响,提高了co-scr反应效率。
7.本发明提供的co-scr脱硝催化剂的制备方法,包括如下步骤;(1)取镁源、钛源制成溶液后混匀,加入分散剂,调节ph为2~4,得到白色凝胶,将凝胶干燥后进行晶化,产物进行洗涤、过滤;(2)配制含步骤(1)产物、柠檬酸的混合液,加入y型分子筛浸渍,取出后干燥、焙烧制得复合载体;(3)将复合载体浸渍到含活性组分前驱体的溶液中,取出后干燥、焙烧,得到催化剂粉体;(4)将催化剂粉体经捏合、陈化、挤出、干燥和焙烧,制得成型催化剂。
8.本发明中,步骤(1)所述的钛源为硫酸钛、钛酸四正丁酯、四氯化钛等中的至少一种,制成溶液的浓度是50~600g/l,优选200~400g/l。
9.本发明中,步骤(1)所述的镁源为氯化镁、碳酸镁、硫酸镁等中的至少一种,制成溶液的浓度是5~20g/l,优选8~15g/l。
10.本发明中,步骤(1)按照镁源与钛源的质量比为1:10~1:50,优选1:20~1:40进行混合。
11.本发明中,步骤(1)所述的分散剂为尿素、柠檬酸等中的至少一种。分散剂与钛源的质量比为1:10~1:50,优选1:20~1:40。
12.本发明中,步骤(1)调节ph采用无机酸,如盐酸、硝酸、硫酸等中的至少一种,调节ph为2~4,得到白色凝胶。将凝胶在100~120℃干燥10~20小时;在300~500℃下晶化6~12小时。
13.本发明中,步骤(2)制得的混合液中,步骤(1)产物的含量为300~4000g/l,柠檬酸的含量为10~300g/l。
14.本发明中,步骤(2)所述y型分子筛的硅铝摩尔比为10~400:1,优选80~300:1。y
型分子筛的添加量为50~400g/l,浸渍时间为10~50min。
15.本发明中,步骤(2)所述干燥温度为80~120℃,干燥时间为1~12h。所述焙烧温度为300~600℃,焙烧时间为1~6h。
16.本发明中,步骤(3)所述的活性组分选自la、ce和sr,活性组分前驱体的溶液中la盐的含量为0.1~2mol/l,ce盐的含量为0.1~2mol/l,sr盐的含量为0.01~0.5mol/l,浸渍时间为5~50min。活性组分的前驱体一般选自它们的可溶性盐。
17.进一步的,在浸渍到含活性组分前驱体的溶液中之前,将复合载体浸渍于草酸铈溶液中,溶液中草酸铈的浓度为2~20g/l,浸渍时间为20~50min,取出后干燥,干燥温度为50~80℃,干燥时间为3~10h,然后再浸渍活性组分。
18.本发明中,步骤(3)所述的干燥温度为40~120℃,干燥时间为3~10h。所述的焙烧温度为300~600℃,焙烧时间为1~10小时。
19.本发明中,步骤(4)所述的成型催化剂采用以下配方,以质量份数计,脱硝催化剂粉体75~90份、玻璃纤维2~6份、木浆0.1~2份、聚丙烯酰胺0.1~2份、羧甲基纤维素0.1~2份、氨水5~10份和去离子水10~40份。其中所述玻璃纤维的直径为10
±
1.5μm,长度5
±
1.5μm,干燥失重(105℃干燥至恒重)≤10%。所述木浆为针叶木浆和阔叶木浆。所述聚丙烯酰胺为阴离子型聚丙烯酰胺,分子量为700~900万。所述羧甲基纤维素的1%水溶液粘度为100~300mpa
·
s,干燥失重(105℃干燥至恒重)≤20%。
20.本发明中,步骤(4)所述的捏合、陈化、挤出、干燥和焙烧等操作均为本领域的常规操作。所述的陈化时间一般为12~48小时。所述干燥温度为20~80℃,干燥时间为1~7天;所述的焙烧温度为300~600℃,焙烧时间为3~20小时。
21.本发明所述的co-scr脱硝催化剂是采用上述本发明方法制备的。所制备的脱硝催化剂中:以催化剂总质量计,y型分子筛含量为5%~20%,tio2含量50%~85%,氧化镁含量为2%~10%,活性组分la含量以金属氧化物计为0.5~10%,ce含量以金属氧化物计0.5~10%,sr含量以金属氧化物计为0.2~2%。
22.进一步的,本发明所述脱硝催化剂中还包括草酸铈,以催化剂总质量计,草酸铈含量为0.2%~2%。
23.本发明所提供的co-scr脱硝催化剂的应用,在氧存在下,在nox、co还原剂和上述脱硝催化剂存在下进行催化脱硝反应,脱硝率可达90%以上。
24.本发明应用中,所述的催化脱硝反应条件为:温度小于500℃,优选300~400℃。
25.与现有技术相比,本发明具有如下优点:(1)本技术发明人在研究过程中发现,通常co-scr脱硝反应会受氧存在的影响,导致co被氧化而脱硝率下降。为了避免氧存在对脱硝反应的影响,发明人采用特定的复合载体及活性金属组分,可以抑制co和o2的反应,提高还原剂co的利用率,提高脱硝效果。
26.(2)采用镁源、钛源制得混合液,并进行ph调控和晶化,随后将y分子筛加入至该混合液中浸渍制得复合载体,有助于增大nox和co的吸附速率和吸附稳定性,同时利于吸附的no和co生成吸附态的中间产物,提高了脱硝效率。
27.(3)通过提高催化剂中的氧空位来提高no吸附可以有效控制co吸附中心和氧空位的利用,la-ce-sr复合氧化物的形成会使ceo2产生更多的氧空位,并提高表面化学吸附氧的比率,从而增强氧迁移率,抑制co和o2转化,提高反应的选择性。
28.(4)在负载活性金属组分前先负载草酸铈,在催化反应中可以增强还原剂效果,进一步提高co脱硝性能。
具体实施方式
29.下面结合具体实施例对本发明方法和效果进行详细说明,实施例在以本发明技术方案为前提下进行实施,给出了详细的实施方式和具体的操作过程,但本发明的保护范围不限于下述的实施例。
30.以下实施例中的实验方法,如无特殊说明,均为本领域常规方法。下述实施例中所用的实验材料,如无特殊说明,均从常规生化试剂商店购买得到。
31.本发明实施例使用的玻璃纤维的直径为10
±
1.5μm,长度5
±
1.5μm,干燥失重(105℃干燥至恒重)≤10%。所述木浆为针叶木浆或阔叶木浆。所述聚丙烯酰胺为阴离子型聚丙烯酰胺,分子量为800万。所述羧甲基纤维素的1%水溶液粘度为200mpa
·
s,干燥失重(105℃时干燥)≤20%。
32.实施例1(1)取10g氯化镁和1l去离子水制成溶液;取200g硫酸钛和1l去离子水制成溶液,混匀,强力搅拌同时加入10g尿素,盐酸调节ph为3,得到白色凝胶停止搅拌;将凝胶110℃干燥16h,在400℃进行晶化10h,产物进行洗涤、过滤。
33.(2)将500g步骤(1)产物和20g柠檬酸加入到1l的去离子水中制成混合液。将60g的y分子筛(硅铝摩尔比为100:1)加入到混合液中进行浸渍,浸渍20min后取出,在100℃干燥2h,400℃焙烧3h,制得复合载体。
34.(3)将复合载体浸渍于含0.6mol/l硝酸镧、0.8mol/l硝酸铈和0.03mol/l硝酸锶的浸渍液中,浸渍时间10min。取出后,在80℃干燥6h,450℃焙烧10h,制得催化剂粉体。
35.(4)将催化剂粉体80份与玻璃纤维6份、木浆0.1份、聚丙烯酰胺0.5份、羧甲基纤维素2份、氨水8份、去离子水20份混合后,经捏合、陈化24h、挤出等工序制得蜂窝体,蜂窝体在70℃条件下干燥3天,在500℃焙烧10h,制得催化剂。
36.催化剂中,以催化剂总质量为基准,tio2含量71%,y分子筛含量11%,氧化镁含量4%,lao2含量2.8%,ceo2含量2.3%,sro含量0.5%,其他物质8.4%。
37.实施例2(1)取6g氯化镁和1l水制成溶液;120g硫酸钛和1l水制成溶液,混匀,搅拌同时加入5g柠檬酸,采用硫酸调节ph为2,得到白色凝胶停止搅拌;将凝胶100℃干燥18h,在350℃晶化12h,洗涤、过滤后得到产物。
38.(2)将400g步骤(1)产物、15g柠檬酸、1l水制成混合液。将80g的y分子筛(硅铝摩尔比为80:1)加入到混合液中进行浸渍,浸渍15min后取出,在90℃干燥3h,350℃焙烧5h,制得复合载体。
39.(3)将复合载体浸渍于含1.2mol/l硝酸镧、1.5mol/l硝酸铈和0.04mol/l硝酸锶的浸渍液中,浸渍时间10min。取出后,在70℃干燥4h,550℃焙烧8h,制得催化剂粉体。
40.(4)将催化剂粉体80份与玻璃纤维5份、木浆0.5份、聚丙烯酰胺0.4份、羧甲基纤维素2份、氨水7份、去离子水20份混合后,经捏合、陈化24h、挤出等工序制得蜂窝体,蜂窝体在60℃下干燥4天,在500℃焙烧9h,制得催化剂。
41.催化剂中,以催化剂总质量为基准,tio2含量67.6%,y分子筛含量11.3%,氧化镁含量5.8%,lao2含量3.1%,ceo2含量5.4%,sro含量0.2%,其他物质6.6%。
42.实施例3(1)取18g氯化镁和1l水制成溶液;310g硫酸钛和1l水制成溶液,混匀,搅拌同时加入8g柠檬酸,采用硝酸调节ph为2,得到白色凝胶停止搅拌;将凝胶120℃干燥10h,在450℃晶化8h,洗涤、过滤后得到产物。
43.(2)将610g步骤(1)产物、15g柠檬酸、1l水制成混合液。将200g的y分子筛(硅铝摩尔比为200:1)加入到混合液中进行浸渍,浸渍15min后取出,在120℃干燥1h,450℃焙烧2h,制得复合载体。
44.(3)将复合载体浸渍于含0.2mol/l硝酸镧、0.3mol/l硝酸铈和0.02mol/l硝酸锶的浸渍液中,浸渍时间10min。取出后,在70℃干燥5h,450℃焙烧10h,制得催化剂粉体。
45.(4)将催化剂粉体86份与玻璃纤维6份、木浆0.1份、聚丙烯酰胺0.5份、羧甲基纤维素2份、氨水9份、去离子水20份混合后,经捏合、陈化24h、挤出等工序制得蜂窝体,蜂窝体在80℃干燥2天。干燥后,在500℃焙烧10h,制得催化剂。
46.催化剂中,以催化剂总质量为基准,tio2含量71.9%,y分子筛含量12.6%,氧化镁含量5.1%,lao2含量1.5%,ceo2含量2.5%,sro含量0.6%,其他物质5.8%。
47.实施例4制备过程及操作条件同实施例1,不同在于:在浸渍到含活性组分前驱体的溶液中之前,将复合载体浸渍于草酸铈溶液中,溶液中草酸铈的浓度为10g/l,浸渍时间为30min,取出后干燥,干燥温度为60℃,干燥时间为6h,然后再浸渍活性组分。催化剂中,以催化剂总质量为基准,tio2含量72.1%,y分子筛含量10.8%,氧化镁含量4.3%,草酸铈含量为2.1%,lao2含量2.9%,ceo2含量3.1%,sro含量0.6%,其他物质4.1%。
48.实施例5制备过程及操作条件同实施例1,不同在于:在浸渍到含活性组分前驱体的溶液中之前,将复合载体浸渍于草酸铈溶液中,溶液中草酸铈的浓度为5g/l,浸渍时间为30min,取出后干燥,干燥温度为60℃,干燥时间为6h,然后再浸渍活性组分。催化剂中,以催化剂总质量为基准,tio2含量72.5%,y分子筛含量10.9%,氧化镁含量4.4%,草酸铈含量为0.9%,lao2含量2.8%,ceo2含量3.0%,sro含量0.6%,其他物质4.9%。
49.实施例6制备过程及操作条件同实施例1,不同在于:在浸渍到含活性组分前驱体的溶液中之前,将复合载体浸渍于草酸铈溶液中,溶液中草酸铈的浓度为18g/l,浸渍时间为30min,取出后干燥,干燥温度为60℃,干燥时间为6h,然后再浸渍活性组分。催化剂中,以催化剂总质量为基准,tio2含量69.1%,y分子筛含量10.5%,氧化镁含量4.6%,草酸铈含量为3.6%,lao2含量2.7%,ceo2含量2.9%,sro含量0.8%,其他物质4.6%。
50.实施例7制备过程及操作条件同实施例1,不同在于:将硫酸钛替换为等摩尔量的钛酸四正丁酯,将氯化镁替换为等摩尔量的碳酸镁,最终制得脱硝催化剂。
51.实施例8制备过程及操作条件同实施例1,不同在于:将硫酸钛替换为等摩尔量的四氯化
钛,将氯化镁替换为等摩尔量的硫酸镁,最终制得脱硝催化剂。
52.比较例1制备过程及操作条件同实施例1,不同在于:步骤(2)采用zsm-5分子筛代替y分子筛。
53.比较例2制备过程及操作条件同实施例1,不同在于:步骤(2)不加入y分子筛。
54.比较例3制备过程及操作条件同实施例1,不同在于:步骤(1)不加入镁源。
55.比较例4制备过程及操作条件同实施例1,不同在于:步骤(1)不加入钛源。
56.比较例5制备过程及操作条件同实施例1,不同在于:步骤(1)活性金属组分采用la和sr,不添加ce。
57.比较例6制备过程及操作条件同实施例1,不同在于:步骤(1)活性金属组分采用ce和sr,不添加la。
58.比较例7制备过程及操作条件同实施例1,不同在于:步骤(1)活性金属组分采用ce和la,不添加sr。
59.测试例1催化剂的脱硝活性测试方法如下:实验装置由配气系统、流量控制(质量流量计)、气体混合器、气体预热器、催化反应器和烟气分析系统构成。将整装蜂窝催化剂(150
×
150
×
1000 mm)切割为小型检测块(45
×
45
×
50mm),然后将小型检测块放入固定管式反应器。测试催化剂co氧化活性时,先通入h2o蒸汽、o2以及载气n2,吸附平衡后再通入co,混合气体总流量304l/h,空速为6000h-1
,co浓度均为200ppm,h2o蒸汽:10%(体积比),o2浓度为2%(体积比),反应温度控制在350℃。测试催化剂co-scr活性时,先通入no、h2o蒸汽、o2以及载气n2,吸附平衡后再通入co,混合气体总流量304l/h,空速为6000h-1
,co浓度均为200ppm,no浓度均为100ppm,h2o蒸汽:10%(体积比),o2浓度为2%(体积比),反应温度控制在350℃。各气体流量由质量流量计控制。气体进入反应器之前先通过气体混合器混合再经过预热器预热。进气口与出气口的co、no、co2、o2浓度由mks傅里叶红外烟气分析仪测定。为了消除表面吸附的影响,系统在通气运行稳定20~30分钟开始采集测试。
60.催化剂的催化活性由co的氧化活性和no的脱硝活性反映,由下式计算:co氧化率=[c
co2(out)
/c
co(in)
]
×
100%。
[0061]
脱硝活性=[(c
0-c)/c0]
×
100%。
[0062]
式中,c0为no初始浓度,c为处理后气体中no浓度。
[0063]
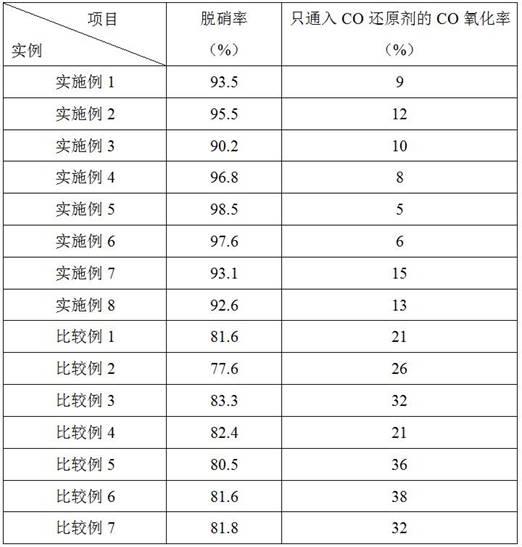
不同实施例和比较例制备的催化剂的co氧化性能、脱硝性能如表1所示。
[0064]
表1
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1