用于分离烃异构体的沸石型吸附剂的制作方法
1.本发明涉及呈包含八面沸石型沸石的附聚物形式的沸石型吸附剂,其用于分离芳族烃的气态或液体混合物,并且更特别地涉及用于分离二甲苯的工艺,并且特别地涉及具有改进的生产率的用于分离间二甲苯的工艺。
2.本发明进一步涉及具有改进的生产率的用于分离异构体的气态或液体混合物的工艺,并且更特别地涉及具有改进的生产率的如下工艺:该工艺用于从包含具有8个碳原子的异构体的芳族烃的进料分离二甲苯的异构体以产生高纯间二甲苯。
背景技术:
3.由y型八面沸石(fau)构成的沸石型吸附剂用于芳族烃混合物中的间二甲苯的选择性吸附的用途是现有技术中熟知的。
4.例如,专利us4306107、us4326092、us5382747、us5900523、和us7728187以及fr2889698和fr2889699显示,包含铝硅酸盐(其含有钠,或者含有钠和锂)的沸石型吸附剂对于分离存在于c8芳族馏分(含有具有8个碳原子的芳族烃的馏分)中的间二甲苯是有效的。专利tw201107032描述了与银或铜交换的沸石y吸附剂对于间二甲苯的收取具有改进的传递和选择性性质。
5.类似于专利us2985589中所描述的那些,专利us5900523中描述的吸附剂用作液相工艺中的吸附试剂,优选地模拟逆流型的工艺的吸附试剂,并且尤其适用于c8芳族馏分。
6.本发明的目的是改进现有的用于制备间二甲苯的工艺并且特别地液相工艺、优选地用于从c8芳族进料分离二甲苯的异构体的模拟逆流型工艺的生产率。出人意料地观察到,通过对用于此类型工艺中的沸石型吸附剂附聚物的特性进行精心选择,可改进所述生产率。
7.本文中,模拟移动床中的分离应以其广义来解释,即,要么其可涉及模拟逆流移动床或模拟并流移动床,要么其可涉及所谓“varicol”工艺。由ludemann-hombourger(o.ludemann-hombourger,r.nicoud,2000)提出并且后来由novasep开发的varicol工艺允许入口和出口管线的非同步位移(bailly等人,2004)。在循环期间,四个区的长度可通过限制实施分离所需的床数而调节。
8.该系列工艺的共同特性是,将沸石型吸附剂附聚物(或更简单地“固体吸附剂”)置于固定床中,并且与所述固体吸附剂接触的液体物流藉由“开-关”阀的组处理,或藉由被称为“旋转阀”的单复合阀处理。
9.当在这些工艺中用作吸附试剂的固体吸附剂的活性要素为沸石时,以晶体形式获得的沸石优选地在工业规模上以附聚物的形式使用。这些以层压体、珠粒或挤出物形式附聚的沸石型吸附剂一般由沸石晶体及粘合剂构成,所述沸石晶体形成就吸附作用而言的活性要素,所述粘合剂旨在确保晶体以附聚物形式凝聚。所述粘合剂也赋予附聚物足够的机械强度以承受其在操作单元中使用时遭受的机械应力。这些机械应力是形成“细粉(fines)”的原因,这些细粉导致整个工艺的操作期间性能劣化。
10.在模拟移动床(smb)中分离二甲苯的工艺已经历许多技术改进,特别是在液体分布器板方面,但是涉及固体吸附剂内在特性的进展相对小。
11.文献us4306107描述了沸石y用作选择性间二甲苯吸附剂的用途,其中可交换的阳离子位点由钠原子占据。为了获得令人满意的间二甲苯选择性,建议使用部分水合的沸石,所述部分水合的沸石具有在950℃下的吸附剂初始重量的2%至7%重量的烧失量(loss on ignition)。在此文献中,建议在模拟移动床工艺中在20℃和250℃之间的温度下并且在大气压和3.5mpa(35巴)之间的压力下进行分离,选择的数值使进料维持液体形式。沸石可交换位点由钠离子占据以及使所述位点活化以获得期望的烧失量允许获得如下的附聚物:其就吸附c8芳族馏分中所含的间二甲苯而言具有改进的容量和选择性性质。
12.用于分离二甲苯的具有对于分离二甲苯而言改进的传递性质的吸附剂在例如国际申请wo2008009845中描述,该申请描述了具有尺寸小于1.7μm的小晶体的沸石x吸附剂,该沸石x吸附剂具有1.15《si/al≤1.5的si/al原子比,与钡交换,并且在国际申请wo2014090771中任选地与钾交换,该申请wo2014090771描述具有特别是对于从c8芳族馏分分离对二甲苯的而言优化的性质的附聚的沸石型吸附剂。这些吸附剂显现出针对对二甲苯选择性及传质的最大性质,同时具有与优化的吸附容量相关的最大机械强度。
13.国际申请wo2018002174提出了呈附聚物形式的沸石型吸附剂,所述沸石型吸附剂具有对于如下而言的优化的性质:分离异构体的气态或液态混合物,更特别地分离气相或液相中的二甲苯,特别地从c8芳族烃馏分分离对二甲苯。该申请的沸石型吸附剂特别显示出针对对二甲苯选择性及传质的最大性质,同时具有改进的强度以及高的吸附容量/吸附剂体积,并且特别适用于液相中对二甲苯的分离工艺,优选地模拟逆流型工艺。
14.特别地,该申请教导,大孔隙率和/或中孔隙率上的强烈增加是不期望的,因而颗粒孔隙率上的强烈增加是不期望的,因为该孔隙率不参与吸附容量。扩散性质和吸附容量的优化是通过特意选择孔隙率和弯曲因子两者而获得的。
15.专利申请us2009/0326308描述了如下的分离工艺:所述工艺使用具有低粘合剂含量并且含有纳米尺寸(典型地小于500nm的平均尺寸)的x型八面沸石晶体的吸附剂。
16.在经由在模拟移动床中的吸附分离二甲苯的工艺中,使沸石型吸附剂与液体进料物流(进料混合物)接触,该进料物流最常包含c8烃的混合物,并且一般且最常包含二甲苯异构体的混合物,更特别地包含邻二甲苯、间二甲苯、对二甲苯、和乙苯的混合物。
17.通过使用含有y型八面沸石结构、与钠交换或者与钠和锂交换的沸石的沸石型吸附剂,间二甲苯相对于进料物流中所有其它烃化合物优先地被吸附在沸石的微孔中。与形成进料物流的初始混合物相比,沸石微孔中的吸附相变得富含间二甲苯。相反地,液相以比表征形成进料物流的初始混合物的相对比例更大的相对比例变得富集诸如邻二甲苯、对二甲苯、和乙苯的化合物。
18.液相从与吸附剂的接触收回(withdrawal),由此形成萃余物流。富集有间二甲苯的吸附相在解吸剂流的作用下解吸,并且从与吸附剂的接触中收回,由此形成萃取物物流。
19.在经由模拟移动床中的吸附进行的二甲苯分离工艺中,固体沸石型吸附剂穿过一或两个多级塔以与液体流接触。多级塔是由沿着基本上竖直(垂直)的轴排列的多个板构成的塔,各板均支撑颗粒状固体的床,并且不同的连续床接收该塔中使用的液体系列的通流(throughflow)。在两个连续床之间存在向各个颗粒状固体的床进料的液体分配装置。
20.一般而言,模拟移动床中的塔的操作可描述如下:
21.塔包含至少四个区,并且任选地包含五个或六个区,这些区的每一个均由一定数量的连续床构成,并且各区均由其在入口管线和出口管线之间的位置定义。典型地,向用于生产间二甲苯的模拟逆流单元(scc)进料至少一种待分馏的进料f(由具有8个碳原子的异构体构成的芳族烃的进料混合物)和有时称为洗脱液(eluent)的解吸剂d,并且从所述单元中收回至少一种萃余物r(含有被选择性地吸附最少的进料产物和解吸剂)以及萃取液e(含有被吸附最多的进料产物和解吸剂)。
22.如例如专利us7208651中所描述,可增加其它入口和出口管线以冲洗分配回路。由于增加这些另外的冲洗流不以任何方式改变scc单元的操作原理,因此为了简洁起见,在本发明的工艺描述中,将不包括这些另外的入口和出口管线。
23.入口和出口管线随时间变化,沿相同方向上移动对应于一个床的数值。如专利us6136198中所教导的,不同入口或出口管线的移动可同时或不同时。根据此第二操作模式的工艺称为varicol。
24.常规上,以模拟逆流(scc)操作的塔中定义了4个不同的色谱区。
25.区1:进料中被吸附最多的产物的解吸区,位于解吸剂d的注入与萃取物e的收回之间。
26.区2:进料中被选择性吸附最少的产物的解吸区,位于萃取物e的收回与待分馏的进料f的注入之间。
27.区3:进料中被吸附最多的产物的吸附区,位于进料的注入与萃余物r的收回之间。
28.区4:位于萃余物r的收回与解吸剂d的注入之间的区。
29.为了增加分离工艺的生产率,现有技术教导,一种方式是改进到沸石型吸附剂附聚物中的整体传递,特别是通过减小晶体的尺寸和/或所述附聚物的平均尺寸。
30.公开了用于药物分离的工艺的现有技术文献(gomes等人,(2006),adsorption,vol.12,p.375sqq.)描述了使用尺寸范围为几十微米直至100μm的附聚物的液相色谱分离工艺。
31.在这些使用非常小尺寸的吸附剂的工艺中,压降δp非常高。对于二甲苯分离工艺,这样水平的压降δp并不常见。然而令人惊讶地观察到压降δp不是尺寸标注标准(dimensioning criterion)。它对吸附器壁的厚度和操作单元的功率有特别的影响。
32.沸石型吸附剂附聚物的另外的特性是所述附聚物的水合速率。通过向进料和/或解吸剂流添加水来确保沸石的水合作用维持在期望的值,例如当沸石y用于通过模拟移动床中的吸附进行的二甲苯分离工艺中时,沸石y的烧失量小于或等于7%。当吸附剂含有沸石fau-y时,针对这样的烧失量水平而添加的水量使得烃流出物(萃取物或萃余物流)中水的重量含量最常介于0ppm和150ppm之间、并且更常介于0ppm和80ppm之间。
33.尽管如此,一般总有必要增加工艺的生产率。
34.所述沸石型吸附剂材料(其为沸石fau-y型的并且特别地含有选自锂、钾、钠、钡、钙及其两种或更多种的混合物的阳离子)可提供用于分离以c8芳族烃馏分中的异构体混合物的形式存在的二甲苯的工艺的显著改进,并且特别地可大大改进c8芳族烃混合物中的间二甲苯的选择性吸附。
35.因此,本发明的第一目的是提供呈附聚物的形式的沸石型吸附剂,其具有对于以
下而言优化的性质:分离异构体的气态或液体混合物,并且更特别地分离气相或液相中的二甲苯,并且特别地从c8芳族馏分分离间二甲苯。特别地,本发明的沸石型吸附剂附聚物展现间二甲苯选择性和传质的最大性质,而同时具有高机械强度和吸附容量,并且特别适用于间二甲苯的液相分离工艺、优选地模拟逆流型工艺中。
36.出于此目的,本发明提出了附聚的吸附剂,优选地八面沸石的附聚的吸附剂,所述吸附剂具有严格地高于1.50、有利地在1.50和6.50之间(排除端点)的si/al原子比,其颗粒孔隙率有利地在25%和45%之间,允许以改进的生产率产生高纯度的间二甲苯,同时避免随着时间的性能劣化。更具体地,本发明涉及沸石型附聚的吸附剂,其包含至少一种具有如下si/al原子比的八面沸石:严格地高于1.50,有利地在1.50和6.50之间(排除端点),优选地在2.00和6.00之间(包括端点),更优选地在2.00和4.00之间(包括端点),并且进一步优选地在2.20和2.80之间(包括端点),并且其中,首先,颗粒孔隙率在25%和45%之间,优选地在30%和45%之间,更优选地在32%和45%之间,进一步优选地在35%和45%之间,并且特别有利地在36%和45%之间,包括端点,以及其次在于,所述附聚物中的晶体尺寸分布的标准偏差σ为小于0.30μm,优选地在0.05μm和0.30μm之间,更优选在0.05μm和0.28μm之间,进一步优选地在0.1μm和0.28μm之间,最优选地在0.1μm和0.25μm之间,包括端点。
技术实现要素:
37.因此,在第一方面中,本发明涉及附聚的沸石型吸附剂,其包含至少一种具有如下的si/al原子比的八面沸石沸石:严格地高于1.50,有利地在1.50和6.50之间(排除端点),优选地在2.00和6.00之间(包括端点),更优选地在2.00和4.00之间(包括端点),并且进一步优选地在2.20和2.80之间(包括端点),特征在于,所述吸附剂的颗粒孔隙率在25%和45%之间,优选地在30%和45%之间,更优选地在32%和45%之间,进一步优选地在35%和45%之间,并且特别有利地在36%和45%之间,包括端点,以及在于所述附聚物中的晶体尺寸分布的标准偏差σ为小于0.30μm,优选地在0.05μm和0.30μm之间,更优选地在0.05μm和0.28μm之间,进一步优选地在0.1μm和0.28μm之间,最优选地在0.1μm和0.25μm之间,包括端点。
38.在一种实施方式中,本发明的附聚的沸石型吸附剂包含具有如下数量加权平均直径的沸石晶体:小于1200nm,优选地在100nm和1200nm之间,更优选地在400nm和1200nm之间,进一步优选地在500nm和1200nm之间,还进一步优选地在550nm和1200nm之间,并且最有利地在600nm和1200nm之间,包括端点。
39.在另一种实施方式中,本发明的附聚的沸石型吸附剂呈具有如下平均直径的珠粒的形式:在100μm和1000μm之间,优选地在100μm和600μm之间,更优选地在200μm和550μm之间,包括端点。
40.在本发明的另一种优选实施方式中,在本发明的附聚的沸石型吸附剂中,通过x射线衍射检测到除所述八面沸石结构以外没有沸石型结构,优选地除八面沸石y结构以外没有沸石型结构。
[0041]“没有(no)”意指,除fau结构以外的沸石结构小于5重量%,优选地小于2重量%,相对于吸附剂的总重量计,重量分数通过xrd确定。
[0042]
此外,优选的是,fau沸石的重量分数、并且优选地fau-y沸石的重量分数高于或等
于80%,相对于本发明的附聚的沸石型吸附剂的总重量计,最高达100%的剩余部分优选地由非沸石相,即基本上对吸附呈惰性的非晶相组成。
[0043]
本发明的附聚的沸石型吸附剂包含一种或多种阳离子,优选地包含碱或碱土阳离子,所述阳离子更优选地选自锂、钠、钡、钙、和钾,单独地或以其两种或更多种的混合物,并且最优选地选自锂、钠、和钙,单独地或以其两种或更多种的混合物。
[0044]
在一种优选的实施方式中,本发明的附聚的沸石型吸附剂中氧化钠(na2o)的含量为按重量计高于5%、更优选地高于8%、进一步优选地高于10%,相对于吸附剂的总重量计,并且有利地,所述氧化钠含量在10%和17%之间、并且典型地在11%和13%之间,包括端点,相对于吸附剂的总重量计。
[0045]
在另一种优选的实施方式中,本发明的附聚的沸石型吸附剂中氧化钾(k2o)的含量为按重量计低于12%,优选地低于5%,更优选地在0和3%之间,进一步优选地在0和2%重量之间,包括端点,相对于吸附剂的总重量计。
[0046]
在另一种优选的实施方式中,本发明的附聚的沸石型吸附剂中氧化钙(cao)的含量为按重量计低于6%,优选地低于5%,更优选地在0和4%之间,进一步优选地在0和3%重量之间,包括端点,相对于吸附剂的总重量计。
[0047]
在本发明的一种实施方式中,所述沸石型吸附剂可具有如下钡含量:低于6%,优选地在0和4%之间,包括端点,以氧化钡bao的重量表示,相对于吸附剂的总重量计。
[0048]
在本发明的一种实施方式中,所述沸石型吸附剂可具有如下锂含量:低于8%,优选地在0和4%之间,包括端点,以氧化锂li2o的重量表示,相对于吸附剂的总重量计。
[0049]
在本发明的另一种实施方式中,除锂、钠、钾、钙、和钡以外的阳离子的总含量一般低于2%,并且最常在0和1%之间,包括端点,此总含量是以所述阳离子的氧化物的重量表示的,相对于沸石型吸附剂的总重量计。
[0050]
在本发明的一实施方式中,本发明的附聚的沸石型吸附剂的烧失量为小于或等于7%,优选地在0和6%之间,更优选地在0和4%之间,进一步优选地在0和3%之间,并且和有利地在0和2.5%之间,包括端点,根据标准nf en 196-2在900℃下测量。
[0051]
在另一方面中,本发明涉及所述附聚的沸石型吸附剂,诸如适才描述的附聚的沸石型吸附剂,在如下工艺中的用途:
[0052]-分离c8芳族异构体馏分,并且特别地二甲苯,并且更特别地间二甲苯;
[0053]-分离经取代的甲苯的异构体,诸如硝基甲苯、二乙基甲苯、甲苯二胺、等等;
[0054]-分离甲苯酚,
[0055]-分离多羟基醇。
[0056]
最后,在进一步方面中,本发明涉及用于在液相或气相中从具有8个碳原子的芳族异构体馏分分离间二甲苯的工艺,所述工艺使用附聚的沸石型吸附剂,诸如此前所定义的并且更具体地本文剩余部分中所定义的,作为间二甲苯吸附试剂。
[0057]
优选地,本发明的用于从具有8个碳原子的芳族烃的异构体馏分分离间二甲苯的工艺是在存在解吸剂的情况下在液相中经由间二甲苯吸附进行的,所述解吸剂可为技术人员已知的任何解吸剂,并且特别地具有低于进料沸点的沸点的任何解吸剂,诸如甲苯或茚满,以及具有高于进料的沸点的沸点的解吸剂,诸如萘满。
[0058]
在一种优选实施方式中,本发明的间二甲苯分离工艺是模拟移动床型的工艺,更
优选地为模拟逆流型的工艺。
具体实施方式
[0059]
优选地,本发明的沸石型吸附剂包含大孔、中孔以及微孔。“大孔”意指具有大于50nm、优选地在50nm和400nm之间的开孔的孔。“中孔”意指具有在2nm和50nm之间(不包括端点)的开孔的孔。“微孔”意指具有小于2nm的开孔的孔。
[0060]
如此前所述,本发明的吸附剂呈具有如下颗粒孔隙率的吸附剂的形式:在25%和45%之间,优选地在30%和45%之间,更优选地在32%和45%之间,进一步优选地在35%和45%之间,并且特别有利地在36%和45%之间,包括端点。此外,所述吸附剂中晶体尺寸分布的标准偏差σ为小于0.30μm,优选地在0.05μm和0.30μm之间,更优选地在0.05μm和0.28μm之间,进一步优选地在0.10μm和0.28μm之间,并且最优选地在0.10μm和0.25μm之间,包括端点。
[0061]
发明人已意料之外地发现,当所述沸石型吸附剂中的晶体尺寸分布的标准偏差σ大于0.3μm时,在间二甲苯分离工艺中、特别是在模拟逆流移动床中进行的液相分离工艺中观察到生产率的急剧下降。
[0062]
还出人意料地观察到,在沸石型吸附剂附聚物中的晶体尺寸分布的标准偏差σ小于0.3μm的情况下,此生产率数值达到最大。此标准偏差σ偏差数值似乎对应于优化的颗粒孔隙率。还完全出人意料地观察到,颗粒孔隙率与沸石型吸附剂附聚物中的晶体尺寸分布的标准偏差σ成反比。因此,标准偏差σ降低地越多,颗粒孔隙率增加地越多。然而,颗粒孔隙率过高导致完全不期望的效应,例如吸附容量损失、机械强度损失,等等。
[0063]
结果是,通过本发明,期望最大生产率的本领域技术人员将寻得颗粒孔隙率和沸石型吸附剂附聚物中的晶体尺寸分布的标准偏差σ之间的折中。因此,通过本发明的附聚的吸附剂,可获得二甲苯分离工艺中、特别是用于分离间二甲苯的二甲苯分离工艺中的最大生产率。
[0064]
有利地,所述附聚的沸石型吸附剂呈具有如下平均直径的珠粒的形式:在100μm和1000μm之间,优选地在100μm和600μm之间,更优选地在200μm和550μm之间,包括端点。
[0065]
优选地,本发明的八面沸石沸石型吸附剂包含钠或锂或钙,或其中两者或三者的混合物。
[0066]
优选地,本发明的沸石型吸附剂含有fau沸石,一般以名称y型沸石表示。“沸石y”意指具有在1.5和6.0之间(排除端点)的si/al原子比的沸石,如此前所述。
[0067]
本发明的沸石型吸附剂附聚物可含有非沸石相(nzp),即基本上对吸附呈惰性的非晶相。本发明的吸附剂的结晶度含量(crystallinity content)(沸石的重量分数)可通过以缩写xrd而为技术人员所知的x射线衍射分析来测量。
[0068]
优选地,本发明的沸石型吸附剂附聚物呈附聚物的形式,即其由至少一种fau沸石(诸如此前所定义的fau沸石)的结晶要素(或晶体)构成,优选地,所述结晶要素(crystalline element)(或更简单地“晶体”)具有如下的数量加权平均直径:小于1200nm,优选地在100nm和1200nm之间,更优选地在400nm和1200nm之间,进一步优选地在500nm和1200nm之间,还进一步优选地在550nm和1200nm之间,并且最有利地在600nm和1200nm之间,包括端点。
[0069]
制备模式
[0070]
本发明的沸石型吸附剂附聚物可通过如下方式制备:适用本领域技术人员已知的操作模式,如例如文献us2009326308、us10125064、tw201107032、和us2007224113中所描述的操作模式,并且选择和调整合成参数,以允许获得的附聚物具有所需的颗粒孔隙率和标准偏差σ的数值。
[0071]
用于合成本发明的沸石型吸附剂附聚物的工艺可例如包含至少以下步骤:
[0072]
a)使以下附聚:至少一种fau-y型沸石的晶体,与包含至少80%的粘土或可沸石化的粘土的混合物的粘合剂,任选地与最高达5%的添加剂,以及与允许形成附聚物材料的量的水;在50℃和150℃之间的温度下将附聚物干燥;将经干燥的附聚物在高于150℃、典型地在180℃和800℃之间、优选地在200℃和650℃之间的温度下在氧化性和/或惰性吹扫气体下煅烧,特别地使用诸如氧气、氮气、空气、干燥和/或经脱碳酸的空气、任选地干燥和/或经脱碳酸的贫氧空气;
[0073]
b)任选地通过使在步骤a)获得的附聚物与碱性基础溶液接触,使全部或部分粘合剂沸石化;
[0074]
c)通过与一种或多种阳离子溶液同时或依次地接触进行步骤a)和/或步骤b)的附聚物的任选的阳离子交换;
[0075]
d)在50℃和150℃之间的温度下,洗涤并且干燥在步骤c)获得的附聚物;以及
[0076]
e)通过加热至一般在100℃和400℃之间、优选地在200℃和300℃之间的温度进行活化,随后收取沸石型附聚的吸附剂。
[0077]
能够在上述合成步骤a)使用的沸石晶体可有利地遵循可在科学文献或专利文献、以及互联网上获得的已知操作模式合成。特别地,所述沸石晶体可如文献us10071914和us2007224113中那样制备。
[0078]
允许对本发明的沸石型吸附剂附聚物的标准偏差σ进行控制的参数例如与在步骤a)所使用的晶体类型相关,特别是与所述晶体的尺寸和标准偏差相关,但还与以下相关:附聚用粘合剂的沸石化条件,例如,温度、时间、碱性沸石化溶液的ph、以及持续时间、搅动模式、剪切速率、压力,等等。
[0079]
更具体地,“在步骤a)所使用的晶体类型”特别地意指例如可通过如下方式控制的所述晶体的标准偏差:调整合成参数,并且特别地合成温度、搅动速度、剪切速率,如例如文献wo2009081022或us2009326308中所述。
[0080]
允许对本发明的沸石型吸附剂附聚物的孔隙率进行控制的合成参数也是技术人员已知的。一般而言,这些参数包含但不限于:粘合剂百分数,附聚物类型(通过挤出、雾化、造粒,等等),湿度水平,粘合剂类型,沸石化条件(温度、时间、碱性沸石化溶液的ph、持续时间、搅动模式、剪切速率、压力,等等)。
[0081]
在一种优选实施方式中,本发明的沸石型吸附剂附聚物的合成不包含添加孔形成剂,孔形成剂的存在可尤其导致结晶度的劣化。
[0082]
还可通过如下方式来制备所述结晶要素:经由晶种合成,和/或调整合成操作条件,诸如sio2/al2o3比率、钠含量、以及合成混合物的碱度。
[0083]
fau型沸石的合成一般是在钠介质(na
+
阳离子)中进行的。如此获得的fau沸石的结晶要素在大部分情况下甚至排它地(仅)含有钠阳离子。然而,使用在以钠形式进行的合
成之间经历一次或多次阳离子交换的结晶要素仍落在本发明的范围内。
[0084]
在步骤a)所使用的fau沸石晶体以及本发明的附聚物中的fau沸石的结晶要素的尺寸是在扫描电子显微镜(sem)下测量的。如此前所述,优选地,晶体的平均直径一般为小于1200nm,优选地在100nm和1200nm之间,更优选地在400nm和1200nm之间,进一步优选地在500nm和1200nm之间,还进一步优选地在550nm和1200nm之间,并且最有利地在600nm和1200nm之间,包括端点。
[0085]
此sem观察还允许验证附聚物中的非沸石相的存在,例如包含残留粘合剂的(在沸石化步骤未转化的)非沸石相,或任何其他无定形相。
[0086]
在本文献中,特别对于结晶沸石要素以及沸石型吸附剂而言,使用术语“数量加权平均直径”或“尺寸”。本说明书中稍后解释这些量级的测量方法。
[0087]
附聚和成形(步骤a)可使用技术人员已知的任何技术进行,诸如挤出,压缩,在盘式造粒器、转鼓造粒器上附聚,雾化,等等。
[0088]
所使用的附聚用粘合剂(见下文定义)和沸石的比例典型地是现有技术中的那些,即5重量份至20重量份粘合剂/95重量份至80重量份沸石。
[0089]
得自步骤a)的附聚物,无论其呈珠粒、挤出物还是其他形式,都一般具有如下的数量加权平均直径或长度(当其为非球形时的最长维度):在0.1mm和1mm之间,并且特别地在0.1mm和0.6mm之间,并且优选地在0.2mm和0.55mm之间,包括端点。
[0090]
在步骤a)之后,例如在挤出物的情形中,最细的附聚物可通过旋风分离器和/或筛分来移除,并且/或过大的附聚物可通过筛分或破碎来移除。
[0091]
本发明的沸石型附聚物材料中所包括的粘合剂包含粘土或粘土的混合物,并且优选地由粘土或粘土的混合物组成。优选地,这些粘土选自:高岭土、高岭石、珍珠陶土、迪开石、埃洛石、凹凸棒石、海泡石、蒙脱石、膨润土、伊利石、和偏高岭土,及其成任何比例的两种或更多种的混合物。
[0092]
对于沸石化步骤,在步骤a)所使用的附聚用粘合剂含有以重量计至少80%、优选地至少90%、更优选地至少95%、更特别地至少96%的至少一种可沸石化的粘土,并且可含有其它矿物粘合剂,诸如膨润土、凹凸棒石,等等。可沸石化的粘土意指能够转化成沸石型材料(最常通过碱性基础溶液的作用实现)的粘土或粘土的混合物。一般而言,所述可沸石化的粘土属于高岭土系列(例如高岭石、珍珠陶土、迪开石、埃洛石)和/或偏高岭土。
[0093]
在任选地于步骤a)所使用的添加剂中,其可包括沸石合成的技术人员、专家已知的任何类型的氧化硅源,例如胶态氧化硅、硅藻土、珍珠岩、粉煤灰、砂、或任何其它形式的固体氧化硅。
[0094]
在步骤a),除fau沸石的结晶要素和粘合剂以外,还可使用例如旨在促进附聚或改进硬化的添加剂,以及技术人员已知的其它添加剂。
[0095]
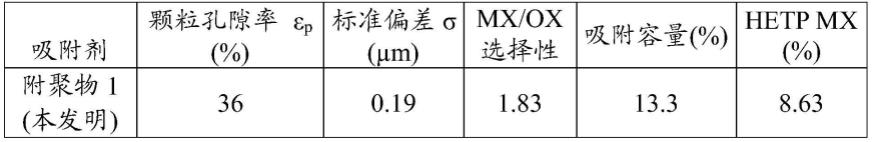
特别地,若所述附聚用粘合剂含有一种或多种可沸石化的粘土,则煅烧允许将可沸石化的粘土、典型地高岭土转化成偏高岭土,偏高岭土可随后在沸石化步骤(步骤b))转化成沸石。其原理在d.w.breck的“zeolite molecular sieves”,john wiley and sons,new york,(1973),314-315页中阐述。
[0096]
附聚用粘合剂的任选的沸石化是使用技术人员目前所熟知的任何方法进行的,并且例如可通过如下方式进行:将得自步骤a)的产物浸入碱性基础溶液中,所述碱性基础溶
液一般是水溶液,例如氢氧化钠和/或氢氧化钾的水溶液。
[0097]
作为一般规律,碱性沸石化溶液的浓度优选地在0.5m和5m之间。优选地,沸石化是在高于环境温度的温度下、并且典型地在80℃至100℃区间内的温度下加热而进行的。沸石化过程(工艺)的持续时间一般在几十分钟和几小时之间,优选地在约1小时和8小时之间。
[0098]
优选地,为了在不使存在的沸石晶体的结晶度劣化的情况下确保所述粘合剂完全沸石化,优选使所述吸附剂与冷氢氧化钠溶液接触,并施加逐步升温,直至80℃-100℃的温度。
[0099]
类似地,氢氧化钠的浓度可维持在相同浓度或其可逐步增加,以维持初始晶体的最大结晶度并且确保可沸石化的粘合剂的最大转化。
[0100]
任选的阳离子交换步骤是根据技术人员已知的常规方法进行的,并且最常通过如下方式进行:使得自步骤a)的附聚物与对应于所需的阳离子交换的阳离子的盐接触。一般而言,所述阳离子交换操作是在环境温度和100℃之间的温度下、优选地在80℃和100℃之间的温度下在水溶液中进行的。
[0101]
优选的是如下操作:使用相对于期望进行交换的沸石的阳离子而言大量摩尔过量的期望的阳离子的水溶液,该操作有利地经由连续交换进行,以获得期望的交换率。
[0102]
在阳离子交换步骤之后,一般进行洗涤(并且任选地使用水),之后干燥如此获得的附聚物。
[0103]
跟随在干燥之后的活化是使用技术人员已知的方法以常规方式进行的,例如,一般在100℃和400℃之间、优选地200℃和300℃之间的温度下,持续根据所需的水含量和烧失量而确定的时间,典型地持续1小时至6小时。
[0104]
表征技术
[0105]
沸石晶体的颗粒尺寸测量:
[0106]
在步骤a)所使用的fau型沸石的要素(即晶体)以及附聚物中所含的沸石y的要素(即晶体)的数量加权平均直径的估计通过在扫描电子显微镜(sem)下观察进行。
[0107]
为了估计样品中沸石颗粒(即晶体)的尺寸,以至少5000倍的放大率拍摄一组图像。随后,使用专门的软件,例如logrami的smile view软件,对至少200个颗粒进行直径测量。精度在3%的区间内。由所述直径测量形成的直方图的测量允许同时确定其分布的标准偏差。
[0108]
沸石型吸附剂附聚物的化学分析——si/al比率和交换率:
[0109]
此前所描述的在步骤a)至e)之后获得的最终产物的元素化学分析可使用技术人员已知的不同分析技术实施。在这些技术中,可提及:在波长色散x射线荧光光谱仪(wdxrf)上,例如bruker的tiger s8上进行的x射线荧光化学分析,诸如标准nf en iso 12677:2011中所描述的。
[0110]
x射线荧光是使用原子在x射线域中的光致发光进行的用于确定样品的元素组成的非破坏性光谱技术。一般通过x射线束或电子轰击实现的原子的激发,在原子回到基态之后生成特定辐射。x射线荧光光谱具有如下优点:其几乎不取决于元素的化学结合,这提供了定性和定量两者的精确确定。在校准之后,对于各氧化物所获得的测量不确定度常规地小于0.4重量%。
[0111]
这些元素化学分析允许验证附聚物中所使用的沸石的si/al原子比和此前所描述
的步骤a)至e)之后获得的最终产物的si/al原子比两者,以及验证在步骤c)描述的离子交换的品质。在本发明的描述中,si/al原子比的测量不确定度为
±
5%。
[0112]
离子交换的品质与交换之后仍在所述沸石型附聚物中的氧化钠(na2o)摩尔数有关。更具体地,阳离子交换率是通过评估所述阳离子的氧化物的摩尔数与存在的阳离子的所有其它氧化物的摩尔数之间的比率来评估。
[0113]
例如,对于含有钠和锂阳离子的沸石型吸附剂附聚物,锂阳离子的交换率是通过评估氧化锂li2o的摩尔数和组(li2o+na2o)的摩尔数之间的比率来评估的。
[0114]
应注意,不同氧化物的含量是以相对于无水沸石型吸附剂的总重量计的重量分数给出的。
[0115]
沸石型吸附剂的颗粒尺寸测量:
[0116]
确定在附聚和形成步骤a)之后获得的沸石型吸附剂的数量加权平均直径通过如下方式实施:根据标准iso 13322
–
2:2006通过成像分析附聚物样品中的颗粒尺寸分布,使用输送器以允许样品在相机前通过。
[0117]
随后,通过应用标准iso 9276-2:2001,由颗粒尺寸分布计算数量加权平均直径。在本文献中,对于沸石型附聚物,使用术语“数量加权平均直径”或者“尺寸”。对于本发明的附聚物的尺寸范围,精度在0.01mm的区间内。
[0118]
沸石型吸附剂的机械强度:
[0119]
诸如本发明中所描述的沸石型吸附剂的床的抗碎强度是通过如下表征的:shell method series sms1471-74“determination of bulk crushing strength of catalysts.compression-sieve method”,与vinci technologies销售的“bcs tester”设备相关。此方法最初旨在表征3mm至6mm的催化剂,基于使用425μm筛网,以允许在破碎时分离细粉(fine)。使用425μm筛网仍适用于直径大于1.6mm的颗粒,但必须顺应寻求表征的附聚物的颗粒尺寸。
[0120]
本发明的附聚物一般呈珠粒或挤出物的形式,一般具有如下的数量加权平均直径或长度(即,对于非球形附聚物而言的最长为敌):在0.2mm和2mm之间、并且特别地在0.1mm和1.0mm之间,优选地在0.1mm和0.6mm之间,包括端点。因此,使用100μm筛网而不是shell标准sms1471-74中提及的425μm筛网。
[0121]
测量方案如下:将附聚的吸附剂的20cm3样品预先用适配的筛网(100μm)筛分,并且预先在250℃(而不是shell标准sms1471-74中提及的300℃)下烘箱干燥至少2小时,置于内部横截面已知的金属筒中。经由活塞通过5cm3不锈钢珠粒的床以增量阶段的方式对此样品施加增加的力,以使通过活塞对吸附剂附聚物施加的力的分布更佳(对于具有严格地小于1.6mm直径的球形颗粒,使用直径2mm的珠粒)。通过筛分分离(适配100μm筛网)在施加压力的不同增量阶段获得的细粉并称重。
[0122]
床内抗碎强度是通过以兆帕(mpa)计的压力测定,在该压力下通过筛网的累积细粉的量为样品的0.5重量%。该值是通过如下方式获得的:将得到的细粉的质量作为施加到吸附剂床上的力的函数绘制在图上,并且内插0.5重量%的累积细粉。床内抗碎强度典型地在几百kpa与几十mpa之间,并且一般为0.3mpa与3.2mpa之间。精度常规地小于0.1mpa。
[0123]
沸石型吸附剂的非沸石相:
[0124]
非沸石相nzp例如沸石化后残留的未沸石化的粘合剂或任何其它无定形相的百分
数是通过以下等式计算的:
[0125]
nzp=100-σ(zp)
[0126]
其中zp表示本发明意义上的沸石y馏分的量的总和。
[0127]
沸石y馏分的百分数(结晶度百分数)是通过以缩写xyd为技术人员所知的x-射线衍射分析测量的。此分析在bruker设备上进行并且使用bruker的topas软件评估沸石y馏分的百分数。
[0128]
微孔体积:
[0129]
附聚物的结晶度也可通过测量其微孔体积将其微孔体积与适当的参考物(在相同的阳离子处理条件下100%结晶沸石,或理论上的沸石)比较来评估。此微孔体积是由气体吸附等温线(例如氮)在该气体液化温度下的测量确定的。
[0130]
在吸附之前,将沸石型吸附剂在真空(p《6.7x10-4
pa)中在300℃和450℃之间的温度下脱气,持续9小时和16小时之间的时间。然后在micromeritics的asap 2020m型设备上进行在77k下的氮吸附等温线的测量,在其中p/p0比率在0.002和1之间的相对压力下取至少35个测量点。
[0131]
大孔和中孔的总体积:
[0132]
大孔vma和中孔vme的体积、颗粒密度dp以及大孔和中孔型的孔隙率ε
p
是通过压汞孔隙测量法测量的。micromeritics的型汞孔隙计用于分析大孔和中孔的孔体积分布。
[0133]
设备操作手册中参照标准astm d4284-83所描述的实验方法由以下组成:将烧失量已知并预先称重的吸附剂样品(待测的附聚物形式的吸附剂)置于孔隙计单元中,并且在在先脱气(30μm汞柱的抽空压力,持续至少10分钟)后,在指定压力(0.0036mpa)下以汞填充所述单元,然后施加不断增大水平的压力直至400mpa,使汞逐渐进入样品的多孔网状结构,使用至少15个压力水平,最高达0.2mpa,随后施加0.1mpa的增量直至1mpa,随后施加0.5mpa的增量直至10mpa,随后施加2mpa的增量直至30mpa,随后施加5mpa的增量直至180mpa,最后施加10mpa的增量直至400mpa。
[0134]
所施加的压力与孔进入阈值的特征尺寸(对应于表观孔径)之间的关系是如下确定的:使用laplace-young方程式,并且假设圆柱形孔开孔、汞和孔壁之间的接触角为140
°
,并且汞表面张力为485dynes cm-1
。记录在各压力水平pi下插入的汞的体积增量δvi,允许随后将所插入的汞的累积体积作为所施加压力v(pi)的函数或作为孔的表观直径v(li)的函数进行绘图。将汞填充所有颗粒间空隙时及填充之后的值设为0.2mpa,并且认为高于此值汞便进入吸附剂的孔中。随后通过如下方式计算颗粒体积vp:孔隙计单元的体积减去在此压力(0.2mpa)下的汞的累积体积,然后将该差值除以等效无水吸附剂的质量,即对烧失量校正过的所述材料的质量。颗粒密度dp是此前定义的颗粒体积vp的倒数。
[0135]
吸附剂的大孔体积vma定义为在0.2mpa与30mpa之间的压力下插入的汞的累积体积,对应于表观直径大于50nm的孔中含有的体积。吸附剂的中孔体积vme定义为在30mpa与400mpa之间的压力下插入的汞的累积体积。由于经由汞压入法测量孔体积的方法无法获得微孔体积,因此诸如通过汞压入法测量的总孔体积vtot对应于大孔体积vma和中孔vme体积的总和。
[0136]
因此,在本文献中,沸石型吸附剂的大孔体积vma和中孔体积vme及其总和(总孔体
积vtot),以cm3g-1
表示,是通过压汞孔隙测量法测量的并且与以无水当量表示的样品质量(即对烧失量校正过的所述吸附剂的质量)有关。颗粒密度dp以gcm-3
表示并且指的是以无水当量计的样品质量。
[0137]
大孔和中孔型颗粒孔隙率ε
p
是颗粒密度dp乘以大孔体积vma和中孔体积vme的总和的积:
[0138]
εp=dp x(vma+vme)
[0139]
沸石型吸附剂的烧失量:
[0140]
烧失量如下确定:遵循标准nf en 196-2(2006年4月)中描述的操作模式,通过在900℃
±
25℃的温度下于空气中煅烧样品,在氧化性气氛中确定。测量的标准偏差小于0.1%。
[0141]
实施例
[0142]
实施例1:制备附聚物
[0143]
制备了两种附聚的吸附剂:附聚物1(根据本发明),以及附聚物2,作为对比例。两种附聚物均如下文所示由nay八面沸石型沸石晶体(其平均尺寸为0.5μm)制备。这些晶体的标准偏差分别为0.23μm和0.60μm。
[0144]
制备附聚物1(根据本发明)
[0145]
将800g的nay沸石晶体(标准偏差0.23μm)、160g高岭土粘土(表示为煅烧当量)、以及195g的商业胶态氧化硅klebosol
tm
30n50(含有30重量%sio2和0.5重量%na2o)均匀混合,并用允许混合物挤出的水进行附聚。将挤出物干燥并在氮气流下于550℃下煅烧(烧制粘土)2小时,随后研磨以获得数均直径等于0.5mm的附聚物。
[0146]
将如此获得的附聚物(20g)置于双夹套玻璃反应器中,并且维持在90℃
±
1℃的温度,随后添加275ml的1.0m氢氧化钠水溶液,并在该温度下将反应介质搅动3.5小时。
[0147]
最后,将附聚物连续用水洗涤3次,随后将反应器排空。通过控制洗涤水的最终ph(其在10.0和10.5之间)来评估洗涤效率。
[0148]
随后,将附聚物在80℃下干燥2小时,并随后在氮气流下在400℃下活化2小时
[0149]
制备附聚物2(对比实施例)
[0150]
将800g的nay沸石晶体(标准偏差0.60μm)、145g高岭土粘土(表示为煅烧当量)、以及180g的商业胶态氧化硅klebosol
tm
30n50(含有30重量%sio2和0.5重量%na2o)均匀混合,并且用使得可将混合物挤出的水进行附聚。将挤出物干燥,并在氮气流下在550℃下煅烧(烧制粘土)2小时,并且随后将其研磨,以获得数均直径等于0.5mm的附聚物。
[0151]
将如此获得的附聚物(20g)置于双夹套玻璃反应器中,并且维持在95℃
±
1℃的温度下,随后添加275ml的1.15m氢氧化钠水溶液,并在此温度下将反应介质搅动3小时。
[0152]
最后,将附聚物连续用水洗涤3次,并将反应器排空。通过控制洗涤水的最终ph(其在10.0和10.5之间)来评价洗涤效率。
[0153]
随后,将附聚物在80℃下干燥2小时,并随后在氮气流下在400℃下活化2小时。
[0154]
附聚物1和附聚物2的特性
[0155]
对于附聚物1和附聚物2,颗粒孔隙率εp的数值以及最终附聚物中晶体的标准偏差σ与以下表1中所示。
[0156]
最终附聚物1中测量的nay晶体的平均尺寸为0.71μm,以及最终附聚物2中测量的
nay晶体的平均尺寸为0.65μm。
[0157]
烧失量是如上文所描述地测量的,并且为2,5%
±
0,1%。
[0158]
实施例2:使用附聚物1和附聚物2进行的突破性测试
[0159]
使用附聚物1和附聚物2进行突破性测试(迎头色谱)。所提及的分析允许评价附聚物各自的分离效率。用于各测试的附聚物量为约26g。
[0160]
突破性曲线是根据以下方法获得的:
[0161]-用附聚物填充柱,并将该柱设置在装置中;
[0162]-在环境温度下,用溶剂填充该柱;
[0163]-在溶剂流(5cm3.min-1
)下逐渐加热,直至达到吸附温度;
[0164]-一旦达到吸附温度,就将溶剂注入设定在30cm3.min-1
的速率。
[0165]-由溶剂切换至进料管线,以注入进料流(30cm3.min-1
);
[0166]-维持进料流注入足够长的时间,以达到热力学平衡(即,直至流出物中溶剂浓度为0);
[0167]
收集并分析流出物。
[0168]
所使用的溶剂为对二乙苯。进料由以下组成:
[0169]
间二甲苯:45重量%
[0170]
邻二甲苯:45重量%
[0171]
异辛烷:10重量%(从分离角度来看惰性化合物在此仅用于示踪目的,以估计非选择性体积)
[0172]
测试是在140℃下进行的。压力水平足够高以避免流体气化,即1mpa。在实验温度下,流体流动的表面速度(流速/柱截面)对于所有实验而言均为约1.2cm.s-1
。
[0173]
间二甲苯(mx)和邻二甲苯(ox)之间的选择性(αmx/ox)由化合物mx和ox的吸附体积qmx和qox(这些由通过柱流出物的分析建立的质量平衡确定)以及入口进料组成计算,其中化合物的体积分数为ymx和yox:
[0174][0175]
获得的结果收集与以下表1中:
[0176]
‑‑
表1
‑‑
[0177][0178][0179]
要点:
[0180]
吸附容量以%表示(对于100cm3的柱而言吸收的c8-芳族化合物的cm3)
[0181]
hetp(height equivalent to a theoretical plate)=对间二甲苯测量的理论塔板高度(等板高度)(表示为柱高度的%)
[0182]
mx=间二甲苯;ox=邻二甲苯
[0183]
这些实验的结果显示,使用根据本发明的附聚的吸附剂允许使对间二甲苯测量的理论塔板高度急剧降低。这清楚地表明了传质容量的改进。分离工艺生产率与传质容量改进直接成正比。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1