采用化学沉淀法制备棕榈酸/二氧化硅相变微胶囊材料的工艺
1.本发明涉及相变材料制备技术领域,尤其涉及采用化学沉淀法制备棕榈酸/二氧化硅相变微胶囊材料的工艺。
背景技术:
2.相变材料(phase change materials,pcms),又称为相变储能材料,它能够根据周围环境的温度变化而作出响应,吸收外界中的热量或向外界放出热量而产生相转变,从而起到控制和调节周围环境温度的作用。目前我国建筑物平均热负荷指标偏高(30
‑
50w/m2),是北欧国家相同气候条件下建筑供暖能耗的2
‑
3倍,建筑供热系统节能的潜力很大。太阳能是一种资源潜力巨大的可再生清洁能源,常呈现“夏盈冬亏”的局面,若使得其能连续地向用户供应能量,储热是一个重要环节。与传统的显热储热相比,相变储热以其单位体积储热量大,储释热过程中温度变化小等优点,在太阳能供热系统等节能领域应用已经备受重视。相变微胶囊材料是将固体或液体相变材料用其他有机或无机材料包覆起来形成的微/纳米颗粒,由于相变材料被一层囊壁包覆,因而使用过程中产生的液漏、相分离以及腐蚀性等问题便可得到有效解决。
3.太阳能供热系统用相变储热材料,不仅要求具有高储释热性还能在快速热冲击下仍保持良好的形态稳定性。由于太阳能供热系统内流体的温度一般在50
‑
80℃左右,正十二醇、棕榈酸、硬脂酸等长链脂肪酸材料的相变温度恰巧也在该范围,此外它们还具有储热密度大,热及化学稳定性良好,可有效克服相变颗粒的分层及凝聚问题等优点。其中棕榈酸pa的相变潜热较大(熔融焓约为200j/g)、价格低廉且易获得,且含有羧基基团,与无机物基体的相容性好。无机壳材能较好的解决有机壳材的力学性能和热稳定性较低的缺点,研究最多的二氧化硅、二氧化钛和碳酸钙等,利用可控沉积方式使无机胶体颗粒逐步沉降在芯材粒子表面,层层包裹形成相变微胶囊材料,具有较好的发展和应用前景。
4.尉菁华等以pa为芯材、正硅酸乙酯teos为硅源,原位缩合制备具有核/壳结构的pa/sio2相变微胶囊,实验表明微胶囊化结构使芯材pa的热稳定性能显著提高。chen等采用溶胶
‑
凝胶法以甲基三乙氧基硅烷(mtes)为硅源制备sio2壳材,石蜡芯材合成相变微胶囊,测试表征显示该相变材料的包裹率达80%以上,相变潜热和热稳定性均较高。罗瑞涟等采用更为环保便宜的九水合硅酸钠为硅源,石蜡为芯材,氯化铵为沉淀剂,成功制得了具有球状结构的paraffin@sio2相变微胶囊材料,其储热能力和热稳定性均较好。pour等采用自组装方法制备了以棕榈酸(pa)为芯材、碳酸铜为壳材的微囊化相变材料,合成的微胶囊表面粗糙,且平均直径较大为1.5
‑
2μm,pa@cuco3相变材料的热稳定性良好。现有专利申请2017106869230《一种二氧化硅包覆的相变微胶囊及其制备方法和应用》公开了相变微胶囊采用溶胶
‑
凝胶反应制备,制得的相变微胶囊平均粒径0.1
‑
100微米也相对较大。
5.可见,采用sio2为壁材的相变材料研究方法较为成熟,其中溶胶凝胶法最为常见。化学沉淀法作为相变材料制备方法的选择,相比于溶胶凝胶法,制备时间短,工艺简单,具
有一定优势;但是,化学沉淀法由于需要沉积的速度和反应速度比较快,很容易出现团聚问题,工艺因素要求较高,此外,化学沉淀法在进行壁材包裹芯材的制备中,由于需要层层包裹,对包裹速率的均匀性要求高,往往不好包裹,包裹效率低,难以做到充分包裹,导致最终制得的相变材料粒径大小不一,获得目标产物品质低,对工艺条件和制备操作提出了更高要求。
6.目前尚未有采用化学沉淀法进行二氧化硅作为壁材、包裹棕榈酸的报道,原因可能在于棕榈酸属于阴离子系列的表面活性剂,沉淀剂尤其盐酸、氯化铵属于强电解质,会对棕榈酸产生影响,使其变粘稠而难以包裹,因此很容易导致相变微胶囊材料制备的失败;即当芯材质量较高、在滴硅酸钠时,会早早出现絮凝现象,也是由于棕榈酸常用作表面活性剂,其水溶液中存在胶团化作用,当棕榈酸浓度过大时,这种作用较为凸显,加入硅酸钠等强电解质,会使胶团双电层压缩,减少表面活性离子间的相互排斥作用,加速了胶团的凝聚,从而使溶液变粘稠,不易搅拌;对工艺提出了较高的要求。
技术实现要素:
7.本发明为了弥补现有技术的不足,提供了采用化学沉淀法制备棕榈酸/二氧化硅相变微胶囊材料的工艺,以棕榈酸(pa)为芯材、无机sio2为壁材,经化学沉淀工艺优化,制备高品质相变微胶囊储能材料,旨在制备能满足太阳能供热系统用的热稳定性良好、高储释热的相变微胶囊材料,该种相变材料也可广泛应用于工业及民用各领域的潜热储存和热管理,对于节能降耗有积极作用,解决了现有技术中存在的问题。
8.本发明是通过如下技术方案实现的:
9.采用化学沉淀法制备棕榈酸/二氧化硅相变微胶囊材料的工艺,包括如下操作步骤:
10.(1)配制均化o/w体系
11.a取适量pa水浴加温,至其全部熔融;备用;
12.b向稀盐酸中加入复合乳化剂,磁力搅拌,预乳化处理;然后将预乳化的盐酸溶液快速滴加至步骤a中,继续搅拌、乳化反应,至形成略显灰色的透明o/w体系;
13.(2)分层相变微胶囊溶液制备
14.配制九水合硅酸钠水溶液,一定温度下慢速滴入步骤(1)的o/w体系中,滴加时间不少于2h,边滴加边搅拌;滴加结束后继续搅拌反应一定时间,然后将反应物取出冷却、陈化,形成分层相变微胶囊溶液;
15.(3)洗涤产物
16.进一步地,步骤(1)中稀盐酸、复合乳化剂的总量为pa质量的15%
‑
20%;稀盐酸和复合乳化剂的质量比为1.5:1;稀盐酸浓度为0.8mol/l
‑
1.0mol/l;芯壁材质量比(pa:九水合硅酸钠)为0.8:1
‑
1:1;九水合硅酸钠水溶液的滴速为0.5ml/min。
17.进一步地,稀盐酸浓度为0.8mol/l、芯壁材质量比为1:1。
18.进一步地,步骤(1)a的pa置于烧瓶中、于水浴升温至70℃,使其全部熔融;步骤(1)b的预乳化温度不低于60℃,预乳化时间为12
‑
18min;继续搅拌的搅拌速率为2000rpm、继续乳化时间为30min。
19.进一步地,步骤(1)b的预乳化时间为15min。
20.进一步地,步骤(1)a的复合乳化剂为十六烷基三甲基溴化铵和聚氧乙烯辛基苯酚醚
‑
10o。
21.进一步地,所述十六烷基三甲基溴化铵和聚氧乙烯辛基苯酚醚
‑
10o的质量比为1.5:1。
22.进一步地,步骤(2)九水合硅酸钠水溶液于70℃条件下慢速滴入步骤(1)的o/w体系中;滴加的搅拌速率为200rpm
‑
400rpm;滴加结束后继续搅拌反应1h;反应物取出后室温冷却、陈化24h。
23.进一步的,步骤(2)九水合硅酸钠水溶液采用蠕动泵慢速滴入步骤(1)的o/w体系中。
24.进一步地,步骤(3)洗涤产物操作欧威:采用热乙醇和去离子水的混合液过滤和洗涤产物,重复多次,去除未包裹的pa和其他杂质;将洗涤后的固体于干燥箱中干燥处理,即得。
25.进一步地,热乙醇和去离子水的重量比为2:1;干燥温度为45℃、干燥时间24h。
26.如前所述的采用化学沉淀法制备棕榈酸/二氧化硅相变微胶囊材料的工艺制得的相变微胶囊材料。
27.进一步地,所述相变微胶囊材料的包覆率和储能效率均高于83%;平均粒径200nm。
28.本发明的有益效果:
29.本发明的相变微胶囊材料以pa作为芯材、二氧化硅作为壁材,采用化学沉淀法进行制备,通过对均化o/w体系及分层相变微胶囊溶液制备过程的控制优化,获得的相变微胶囊材料包覆率和储能效率均高于83%、平均粒径200nm,应用范围广泛,使用安全。该制备方法工艺简单,操作方便,o/w体系中盐酸浓度的控制、复合乳化剂的选择、分层相变微胶囊溶液制备时芯壁材质量的控制,以及九水合硅酸钠的滴速的控制,一是保证了二氧化硅壳材的硅酸凝胶的缩聚速度与其胶束液滴在pa表面沉积的速率基本一致,从而使得形成的相变材料壳材光滑致密,包覆率高;二是避免了在滴硅酸钠时,早早出现絮凝现象,且避免了后续结块现象的问题;三是控制好了壁材沉积速度,避免了粒径较大、黏连现象的发生,提升了包裹效率,且便于后续洗涤,最终得到的相变材料形貌较好,大小均匀、分散性佳,热循环稳定性优异。本发明的九水合硅酸钠相比现有技术的其他硅源,如正硅酸乙酯等没有毒性,价格经济。通过前述整体工艺的选择、优化和调整,采用化学沉淀法显著改善了制备的相变微胶囊材料的团聚问题,提高了储能效率,获得了粒径更均一的产品。
附图说明
30.图1为本发明pa@sio2相变微胶囊制备工艺流程图;
31.图2为本发明不同合成条件下pa@sio2相变微胶囊的dsc图;
32.图3为本发明不同条件下制得的pa@sio2相变微胶囊的微观形貌;
33.图4为本发明实施例1的pa@sio2相变微胶囊eds元素定性分析图;
34.图5为纯pa、sio2和本发明实施例1制备pa@sio2相变微胶囊xrd谱图;
35.图6为本发明不同合成条件下pa@sio2相变微胶囊的xrd谱图;
36.图7为纯pa、sio2和本发明实施例1条件下制备pa@sio2的ft
‑
ir图谱;
37.图8为不同合成条件下pa@sio2相变微胶囊的ft
‑
ir谱图;
38.图9为pa和本发明实施例1条件下制备pa@sio2相变微胶囊的tg曲线;
39.图10为本发明实施例1制备pa@sio2相变微胶囊的dsc热循环图;
40.其中,图2中a为pa和实施例1合成条件下pa@sio2相变微胶囊的dsc图;图2中b为实施例2
‑
5合成条件下pa@sio2相变微胶囊的dsc图;图2中c为实施例6
‑
10合成条件下pa@sio2相变微胶囊的dsc图;
41.图3中(a)为实施例1最佳工艺条件下制得;图3中(b)为实施例2盐酸氢离子浓度1.0mol/l条件下制得;图3中(c)为实施例3盐酸浓度1.2mol/l条件下制得;图3中(d)为实施例4芯壁材质量比0.8:1条件下制得;图3中(e)为实施例5芯壁材质量比1.2:1条件下制得;图3中(f)为实施例6九水合硅酸钠滴速0.3ml/min条件下制得;图3中(g)为实施例7九水合硅酸钠滴速1.0ml/min条件下制得;图3中(h)为实施例8只采用乳化剂ctab的条件制得,图3中(i)为实施例9只采用乳化剂op
‑
10的条件制得,图3中(j)为实施例10替换为其他复合乳化剂的条件制得。
具体实施方式
42.为能清楚说明本方案的技术特点,下面通过具体实施方式,结合附图,对本发明进行详细阐述。
43.原材料棕榈酸palmiticacid又称软脂酸pa,97%,相变温度63℃,作为相变材料芯材;偏硅酸钠九水化合物(na2sio3·
9h2o)作为制备壳材的硅源,均由上海麦克林生化有限公司提供。以十六烷基三甲基溴化铵ctab和聚氧乙烯辛基苯酚醚
‑
10(op
‑
10)为复合乳化剂,其中ctab由上海麦克林生化有限公司生产,op
‑
10由天津市凯通化学试剂有限公司生产,纯度≥70%。以37%浓盐酸为沉淀剂,国药集团化学试剂有限公司生产。相变材料后处理溶剂无水乙醇购自天津市凯通化学试剂有限公司。去离子水为实验室自制。
44.图1为采用化学沉淀法制备pa@sio2相变微胶囊材料的示意,二氧化硅壁材由九水合硅酸钠与盐酸反应制得,反应方程式如下:
45.na2sio3·
9h2o+2hcl=h2sio3+2nacl+9h2o
[0046][0047]
实施例1
[0048]
该化学沉淀法制备棕榈酸/二氧化硅相变微胶囊材料的工艺,包括如下操作步骤:
[0049]
(1)配制均化o/w体系
[0050]
a向四口烧瓶中加入11.37g的pa,水浴锅升温至70℃,至其全部熔融;备用;
[0051]
b向0.8mol/l、100ml的稀盐酸中加入复合乳化剂ctab和op
‑
10,其中,乳化剂ctab和op
‑
10的质量比为1.5:1,总量为芯材质量的15%,经磁力搅拌器进行磁力搅拌,60℃以上预乳化处理15min;然后将预乳化的盐酸溶液快速滴加至步骤a的四口烧瓶中,提高搅拌速率至2000rpm,继续乳化30min,至形成略显灰色的透明o/w体系;
[0052]
(2)分层相变微胶囊溶液制备
[0053]
取九水合硅酸钠11.37g,配制九水合硅酸钠水溶液;在70℃条件下,用leadfluidbt102s型号的蠕动泵的细管慢速滴入步骤(1)的o/w体系中,滴加速率为0.5ml/min,滴加时间不少于2h,搅拌速率在300rpm左右;九水合硅酸钠水溶液滴加结束后继续搅
拌反应1h,然后将反应物取出室温下冷却、陈化24h,形成分层的相变微胶囊溶液;
[0054]
(3)洗涤产物
[0055]
采用质量比2:1的热乙醇和去离子水配制混合液,过滤和洗涤产物,重复多次,去除未包裹的pa和其他杂质;将洗涤后的白色固体于45℃干燥箱中干燥处理24h,得到最终产品。
[0056]
实施例2(氢离子浓度为1.0mol/l)
[0057]
该化学沉淀法制备棕榈酸/二氧化硅相变微胶囊材料的工艺,包括如下操作步骤:
[0058]
(1)配制均化o/w体系
[0059]
a向四口烧瓶中加入11.37g的pa,水浴锅升温至70℃,至其全部熔融;备用;
[0060]
b向1.0mol/l、100ml的稀盐酸中加入复合乳化剂ctab和op
‑
10,其中,乳化剂ctab和op
‑
10的质量比为1.5:1,总量为芯材质量的15%,经磁力搅拌器进行磁力搅拌,60℃以上预乳化处理15min;然后将预乳化的盐酸溶液快速滴加至步骤a的四口烧瓶中,提高搅拌速率至2000rpm,继续乳化30min,至形成略显灰色的透明o/w体系;
[0061]
(2)分层相变微胶囊溶液制备
[0062]
取九水合硅酸钠11.37g,配制九水合硅酸钠水溶液;在70℃条件下,用lead fluid bt102s型号的蠕动泵的细管慢速滴入步骤(1)的o/w体系中,滴加速率为0.5ml/min,滴加时间不少于2h,搅拌速率在300rpm左右;九水合硅酸钠水溶液滴加结束后继续搅拌反应1h,然后将反应物取出室温下冷却、陈化24h,形成分层的相变微胶囊溶液;
[0063]
(3)洗涤产物
[0064]
采用质量比2:1的热乙醇和去离子水配制混合液,过滤和洗涤产物,重复多次,去除未包裹的pa和其他杂质;将洗涤后的白色固体于45℃干燥箱中干燥处理24h,得到最终产品。
[0065]
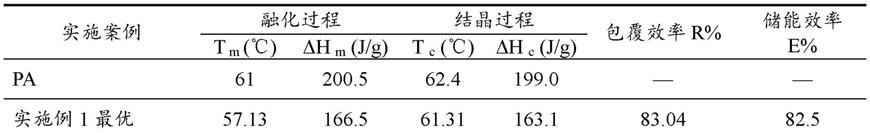
实施例3(氢离子浓度为1.2mol/l)
[0066]
该化学沉淀法制备棕榈酸/二氧化硅相变微胶囊材料的工艺,包括如下操作步骤:
[0067]
(1)配制均化o/w体系
[0068]
a向四口烧瓶中加入11.37g的pa,水浴锅升温至70℃,至其全部熔融;备用;
[0069]
b向1.2mol/l、100ml的稀盐酸中加入复合乳化剂ctab和op
‑
10,其中,乳化剂ctab和op
‑
10的质量比为1.5:1,总量为芯材质量的15%,经磁力搅拌器进行磁力搅拌,60℃以上预乳化处理15min;然后将预乳化的盐酸溶液快速滴加至步骤a的四口烧瓶中,提高搅拌速率至2000rpm,继续乳化30min,至形成略显灰色的透明o/w体系;
[0070]
(2)分层相变微胶囊溶液制备
[0071]
取11.37g的九水合硅酸钠;配制九水合硅酸钠水溶液,在70℃条件下,用lead fluid bt102s型号的蠕动泵的细管慢速滴入步骤(1)的o/w体系中,滴加速率为0.5ml/min,滴加时间不少于2h,搅拌速率在300rpm左右;九水合硅酸钠水溶液滴加结束后继续搅拌反应1h,然后将反应物取出室温下冷却、陈化24h,形成分层的相变微胶囊溶液;
[0072]
(3)洗涤产物
[0073]
采用质量比2:1的热乙醇和去离子水配制混合液,过滤和洗涤产物,重复多次,去除未包裹的pa和其他杂质;将洗涤后的白色固体于45℃干燥箱中干燥处理24h,得到最终产品。
[0074]
实施例4(芯壁材质量比0.8:1)
[0075]
该化学沉淀法制备棕榈酸/二氧化硅相变微胶囊材料的工艺,包括如下操作步骤:
[0076]
(1)配制均化o/w体系
[0077]
a向四口烧瓶中加入9.10g的pa,水浴锅升温至70℃,至其全部熔融;备用;
[0078]
b向0.8mol/l、100ml的稀盐酸中加入复合乳化剂ctab和op
‑
10,其中,乳化剂ctab和op
‑
10的质量比为1.5:1,总量为芯材质量的15%,经磁力搅拌器进行磁力搅拌,60℃以上预乳化处理15min;然后将预乳化的盐酸溶液快速滴加至步骤a的四口烧瓶中,提高搅拌速率至2000rpm,继续乳化30min,至形成略显灰色的透明o/w体系;
[0079]
(2)分层相变微胶囊溶液制备
[0080]
取九水合硅酸钠11.37g,配制九水合硅酸钠水溶液;在70℃条件下,用lead fluid bt102s型号的蠕动泵的细管慢速滴入步骤(1)的o/w体系中,滴加速率为0.5ml/min,滴加时间不少于2h,搅拌速率在300rpm左右;九水合硅酸钠水溶液滴加结束后继续搅拌反应1h,然后将反应物取出室温下冷却、陈化24h,形成分层的相变微胶囊溶液;
[0081]
(3)洗涤产物
[0082]
采用质量比2:1的热乙醇和去离子水配制混合液,过滤和洗涤产物,重复多次,去除未包裹的pa和其他杂质;将洗涤后的白色固体于45℃干燥箱中干燥处理24h,得到最终产品。
[0083]
实施例5(芯壁材1.2:1)
[0084]
该化学沉淀法制备棕榈酸/二氧化硅相变微胶囊材料的工艺,包括如下操作步骤:
[0085]
(1)配制均化o/w体系
[0086]
a向四口烧瓶中加入13.64g的pa,水浴锅升温至70℃,至其全部熔融;备用;
[0087]
b向0.8mol/l、100ml的稀盐酸中加入复合乳化剂ctab和op
‑
10,其中,乳化剂ctab和op
‑
10的质量比为1.5:1,总量为芯材质量的15%,经磁力搅拌器进行磁力搅拌,60℃以上预乳化处理15min;然后将预乳化的盐酸溶液快速滴加至步骤a的四口烧瓶中,提高搅拌速率至2000rpm,继续乳化30min,至形成略显灰色的透明o/w体系;
[0088]
(2)分层相变微胶囊溶液制备
[0089]
取九水合硅酸钠11.37g,配制九水合硅酸钠水溶液;在70℃条件下,用lead fluid bt102s型号的蠕动泵的细管慢速滴入步骤(1)的o/w体系中,滴加速率为0.5ml/min,滴加时间不少于2h,搅拌速率在300rpm左右;九水合硅酸钠水溶液滴加结束后继续搅拌反应1h,然后将反应物取出室温下冷却、陈化24h,形成分层的相变微胶囊溶液;
[0090]
(3)洗涤产物
[0091]
采用质量比2:1的热乙醇和去离子水配制混合液,过滤和洗涤产物,重复多次,去除未包裹的pa和其他杂质;将洗涤后的白色固体于45℃干燥箱中干燥处理24h,得到最终产品。
[0092]
实施例6(九水合硅酸钠滴速0.3)
[0093]
该化学沉淀法制备棕榈酸/二氧化硅相变微胶囊材料的工艺,包括如下操作步骤:
[0094]
(1)配制均化o/w体系
[0095]
a向四口烧瓶中加入11.37g的pa,水浴锅升温至70℃,至其全部熔融;备用;
[0096]
b向0.8mol/l、100ml的稀盐酸中加入复合乳化剂ctab和op
‑
10,其中,乳化剂ctab
bt102s型号的蠕动泵的细管慢速滴入步骤(1)的o/w体系中,滴加速率为0.5ml/min,滴加时间不少于2h,搅拌速率在300rpm左右;九水合硅酸钠水溶液滴加结束后继续搅拌反应1h,然后将反应物取出室温下冷却、陈化24h,形成分层的相变微胶囊溶液;
[0117]
(3)洗涤产物
[0118]
采用质量比2:1的热乙醇和去离子水配制混合液,过滤和洗涤产物,重复多次,去除未包裹的pa和其他杂质;将洗涤后的白色固体于45℃干燥箱中干燥处理24h,得到最终产品。
[0119]
实施例9(替换为单一乳化剂)
[0120]
该化学沉淀法制备棕榈酸/二氧化硅相变微胶囊材料的工艺,包括如下操作步骤:
[0121]
(1)配制均化o/w体系
[0122]
a向四口烧瓶中加入11.37g的pa,水浴锅升温至70℃,至其全部熔融;备用;
[0123]
b向0.8mol/l、100ml的稀盐酸中加入乳化剂op
‑
10,乳化剂总量为芯材质量的15%,经磁力搅拌器进行磁力搅拌,60℃以上预乳化处理15min;然后将预乳化的盐酸溶液快速滴加至步骤a的四口烧瓶中,提高搅拌速率至2000rpm,继续乳化30min,至形成略显灰色的透明o/w体系;
[0124]
(2)分层相变微胶囊溶液制备
[0125]
取九水合硅酸钠11.37g,配制九水合硅酸钠水溶液;在70℃条件下,用lead fluid bt102s型号的蠕动泵的细管慢速滴入步骤(1)的o/w体系中,滴加速率为0.5ml/min,滴加时间不少于2h,搅拌速率在300rpm左右;九水合硅酸钠水溶液滴加结束后继续搅拌反应1h,然后将反应物取出室温下冷却、陈化24h,形成分层的相变微胶囊溶液;
[0126]
(3)洗涤产物
[0127]
采用质量比2:1的热乙醇和去离子水配制混合液,过滤和洗涤产物,重复多次,去除未包裹的pa和其他杂质;将洗涤后的白色固体于45℃干燥箱中干燥处理24h,得到最终产品。
[0128]
实施例10(替换为其他复合乳化剂)
[0129]
该化学沉淀法制备棕榈酸/二氧化硅相变微胶囊材料的工艺同实施例1的制备工艺,所不同的是,所述复合乳化剂替换为十二烷基硫酸钠sds和脂肪醇聚氧乙烯醚aeo
‑
5。
[0130]
一、性能表征
[0131]
1.1采用德国netzsch公司200f3型号的差示扫描量热仪测试微胶囊的初始相变温度、峰值相变温度、热焓等性能。通过下面的公式计算合成相变微胶囊的包裹效率和储能效率等;并通过热循环法测定相变微胶囊的热耐久性。测试条件为在n2气氛下,称取3
‑
5mg粉末样品于固体坩埚中,与参比样对比;以10℃/min速率先降温至30℃,再以相同速率升温至100℃,最后降温至0℃。
[0132]
相变微胶囊的包覆率r
[0133]
相变微胶囊的储能效率e
[0134]
其中式中:δh
m,mpcms
和δh
c,mpcms
分别表示制备相变微胶囊材料mpcms的熔化焓值和
凝固焓值;δh
m,pa
和δh
c,pa
分别表示芯材pa的熔化焓值和凝固焓值。
[0135]
1.2采用德国布鲁克公司tensor37傅里叶变换红外光谱(ftir),采用压片法表征合成相变微胶囊的化学结构特征,分析芯壁材间有无发生化学反应。
[0136]
1.3 x射线衍射分析采用德国布鲁克公司brukerd8x,测试管电压为40kv,管电流为30ma,cu靶,测试角度范围10
‑
60
°
,表征合成相变微胶囊材料的矿物组成。
[0137]
1.4采用德国netzsch公司tg209f3热重分析仪测试合成的相变微胶囊的热稳定性。测试条件为:n2气氛下,室温到600
°
с,流量100ml/min,升温速率10℃/min。
[0138]
1.5导热系数采用瑞典hotdisktps2500s导热系数仪,参照iso22007
‑
2标准瞬态平板热源法进行测试,测试温度选择20℃和65℃两个温度点。
[0139]
1.6利用日本jeol热场发射扫描电子显微镜jsm
‑
7200f观察合成的相变微胶囊材料的形貌和平均的粒径,测试前需要将试样进行真空镀金并粘贴在导电胶带上待测。
[0140]
二、结果与分析
[0141]
2.1 pa@sio2相变微胶囊的合成条件
[0142]
实施例1为最优工艺制得的pa@sio2相变微胶囊;实施例2和实施例3为调整o/w体系中稀盐酸浓度的工艺改变制得的pa@sio2相变微胶囊;实施例4和实施例5为调整芯壁材质量比的工艺改变制得的pa@sio2相变微胶囊;实施例6和实施例7为改变九水合硅酸钠溶液滴速制得的pa@sio2相变微胶囊;实施例8和实施例9为将复合乳化剂替换为单一乳化剂制得的pa@sio2相变微胶囊,实施例10为将复合乳化剂替换为其他复合乳化剂制得的pa@sio2相变微胶囊。
[0143]
表1 pa和不同条件制备的相变微胶囊的相变性能
[0144][0145][0146]
由表1中的数据可以看出,与纯pa的性能相比,各实施例不同条件下制备的相变微胶囊的tm起融温度较棕榈酸有所提前,这说明二氧化硅可以有效的提高微胶囊的导热率,使热量提前传输到芯材发生融化。另相变潜热均有所降低,相变温度范围(tc
‑
tm)较宽,这
是由于无机sio2壳材的包覆减少了pa的含量,二氧化硅壁材限制pa分子的运动导致的。
[0147]
由图2中a、b、c可知,有部分相变微胶囊的吸放热峰不平滑,出现了双峰,这可能是相变微胶囊有些许破损,包裹不严。纯pa的放热和吸热峰均不平滑,这是因为pa在吸放热过程中存在亚稳态的旋转相所致。实施例1制备的pa@sio2微胶囊的吸放热峰为单峰,说明其相变储热性能较好,其过冷度也比纯pa小,这是因为壳材sio2也可作为成核剂,构成的多相成核体系能够降低相变微胶囊溶液过冷度。实施例1和实施例2,当盐酸的c(h
+
)为0.8mol/l、1.0mol/l时,制备sio2壳材的硅酸凝胶的缩聚速率与其胶束液滴在pa表面沉积的速率基本一致,形成的相变材料的壳材较为光滑致密,使其包覆率最高可达83.04%;储能效率在82.5%;但当实施例3的c(h
+
)为1.2mol/l时,盐酸过量会降低硅酸凝胶的缩聚速度,然而胶束的沉积速率会加快,这样形成的壳材厚度较大,且影响其稳定性,包覆率和储能效率在57%左右。当芯壁材质量比变化时,如实施例4芯壁材质量比为0.8:1,合成的相变材料的相变性能较好,包覆率均在74%以上。当芯材质量较高时,芯壁材质量比为1.2:1,如实施例5,在滴硅酸钠时,早早出现了絮凝现象,当将悬浮液倒出后,放置5min后出现结块现象,说明这是温度降低后pa凝固成固态导致。由于硅酸钠合成的二氧化硅壳材粒径较小,过多的芯材,导致包覆需要的壳材量大,形成的壳材较薄,容易破损。
[0148]
实施例6中九水合硅酸钠水溶液的滴速过快,滴速为1.0ml/min时,壁材的沉积速度过快,粒径较大,有粘连现象,包裹效率受到影响,陈化后粘稠状液体中夹杂颗粒和结块物体,不易抽滤。滴速为0.3ml/min,如实施例7,沉淀反应时间近5个小时,生成的壁材速率慢,粒径细小,很多小粒径的微胶囊团聚在一起,形成表面不光滑的较大的球状微胶囊粒子,dsc测试数据显示其相变性能的值也较高。
[0149]
采用复合乳化剂较采用单一乳化剂的乳化效果更显著,制备得到的微胶囊的相变潜热也较高,由于乳化效果决定着芯材能够形成稳定的乳液,从而被壁材包裹,对相变微胶囊的成功制备和相变性能影响巨大。实施例8中,单独使用ctab作为乳化剂时,试样呈现出球形结构,团聚现象严重,小球之间相互连接、层层堆积。实施例9采用单一op
‑
10乳化剂制备的相变微胶囊并没有呈现出密封的胶囊结构,却表现为疏松多孔的块状物体,表面粗糙;其包覆率和储能效率仅在20%左右。实施例10中采用其他复合乳化剂时:十二烷基硫酸钠和脂肪醇聚氧乙烯醚,试样能看到较明显的球形胶囊结构,但团聚现象仍然较为严重。考虑到硅酸水解后溶液呈酸性和负性,因此阳离子表面活性剂效果要优于阴离子表面活性剂。
[0150]
图3中(a)为实施例1最优工艺制得的相变微胶囊的微观形貌。图3中(b)
‑
(j)为实施例2
‑
10制得的相变微胶囊的微观形貌。图3中(a)的相变微胶囊,粒径均匀在200nm左右,且分散性好。实施例2和实施例4对应图3中(b)和(d)中的相变微胶囊相变微胶囊有一定程度的团聚,粒径大小较为均匀,可能是由于相变微胶囊的壁材在后处理时有破损。其他的图中相变微胶囊粒子的团聚程度均较为严重,粒径大小不一,小球之间相互连接、层层堆积,形成了类似与块状的结构。其中实施例9中仅有少量的球形小粒子,没有形成微胶囊的核壳结构,这是因为单独使用非离子表面活性剂的乳化作用效果不好,氢离子没有均匀分布在芯材乳液液滴中,壁材形成受到较大影响。
[0151]
对实施例1制备的pa@sio2相变微胶囊进行x射线能谱分析,图4为相变微胶囊的eds元素定性分析,其主要的元素为c、o和si,其中c元素的含量在72.49%,o元素含量在18.01%;si元素含量在6.74%;还有很少量的na和cl元素。其中主要元素c、o、si的比例与
相变材料芯壁材化合物的组成基本一致。
[0152]
由图5可以看出,棕榈酸pa(jcpds no.24
‑
1853)具有典型的晶体结构,三强峰主要出现在2θ为21.6
°
、24.2
°
和12.4
°
。而sio2则是不定形非晶结构,在20
°‑
27
°
范围有个微微隆起的馒头型衍射峰。实施例1制备的pa@sio2相变微胶囊的xrd图谱则可见是两者的集合,基本呈现了pa的形状,这是由于sio2衍射峰强度低,所以看的不太明显;但pa衍射峰的强度在相变微胶囊中略有降低,这是由于sio2的包覆干扰了pa晶体的生长,导致其结晶峰强度降低。另谱图中没有新的矿物相,这表明制备pa@sio2相变微胶囊的过程中pa的晶体结构为发生改变,这与后面的红外谱图的结论一致。
[0153]
图6为不同实施例条件下制备的pa@sio2相变微胶囊的xrd谱图,由于xrd表征为定性反应,且pa@sio2相变微胶囊的芯壁材不发生反应,只要能够包裹形成核壳结构的微胶囊材料,其xrd谱图就基本相似。从图中能明显看出,pa衍射峰明显的为实施例1、2和4,由于sio2衍射峰强度低,图中几乎看不出来其在20
‑
27
°
范围内的隆峰。实施例9由于包裹率低仅20%左右,所以其xrd的谱图仅显示了sio2壳材的无定形峰。
[0154]
图7中pa在2916cm
‑1和2849cm
‑1处的吸收峰是甲基
‑
ch3和亚甲基
‑
ch2的伸缩振动峰,在1719cm
‑1处则是羧基c=o的伸缩振动峰,位于1295cm
‑1,941cm
‑1和719cm
‑1的吸收峰是由
‑
oh的面内外弯曲振动和旋转振动产生的。sio2谱图中在3441cm
‑1和955cm
‑1的吸收带是si(oh)4溶胶中的
‑
oh的伸缩特征峰和si
‑
oh官能团的弯曲振动峰,位于1078cm
‑1、794cm
‑1处的吸收峰是si
‑
o
‑
si的弯曲振动峰。有pa和sio2的主要吸收峰均出现在pa@sio2相变微胶囊的谱图中,且谱线峰值没有发生偏移,也未出现新的官能团,表明芯材pa与壁材sio2未发生化学反应,仅是进行了物理包覆。
[0155]
图8为不同实施例条件下制备的pa@sio2相变微胶囊的ft
‑
ir谱图,与xrd谱图相似,红外谱图也是一种定性表征的手段,只要能够包裹形成核壳结构的微胶囊材料,其红外谱图就基本相似。不过包裹率越好的,则ft
‑
ir谱图中sio2的si
‑
oh和si
‑
o
‑
si特征官能团以及芯材pa的甲基
‑
ch3和亚甲基
‑
ch2的特征峰较为显著,如实施例1、2、4。其他几个实施案例的谱图中sio2官红能团特征峰不明显,这可能是由于相变微胶囊包裹不严,后期有部分芯材泄露。实施例9由于其包裹率很低,则基本呈现的是sio2的特征峰,以上谱图分析结果与xrd谱图结果基本一致。
[0156]
图9为纯pa和实施例1条件下制备pa@sio2相变微胶囊的tg曲线,图中显示了纯pa在170
‑
260℃范围内有明显的热挥发引起的失重,失重率接近95%。而pa@sio2相变微胶囊有两个失重过程,第一阶段的热失重率较大,约55%左右,失重温度范围在200
‑
280℃,这一阶段主要发生了部分pa从壳材中泄露挥发;第二阶段在350
‑
450℃有个微弱的失重,主要是由于壳材中硅羟基在高温下发生了脱水反应。对比两图可知,由于二氧化硅的热稳定性良好且熔点较高,包裹芯材pa使其不会因为熔化而流失,因此无机二氧化硅壳材的包裹可以极大的提高芯材pa的热稳定性。
[0157]
图10为实施例1条件下制备pa@sio2相变微胶囊经过300次循环升降温的dsc循环曲线图。图中可以明显看出这些循环曲线几乎都重叠在一起,储热性能参数变化不大,说明经过多次吸热放热后微胶囊的性能保持较好,具有较好的热稳定性。这是由于棕榈酸分子羧基间形成的缔合分子对,形成的氢键结合力较强,本身的稳定性较强,外层包裹二氧化硅壳材的热稳定性和熔点更高,能更有效的改善棕榈酸的性能。
[0158]
上述具体实施方式不能作为对本发明保护范围的限制,对于本技术领域的技术人员来说,对本发明实施方式所做出的任何替代改进或变换均落在本发明的保护范围内。
[0159]
本发明未详述之处,均为本技术领域技术人员的公知技术。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1