一种抗生素菌渣资源化处理的方法
1.本发明属于废弃物资源化处理技术领域,特别涉及一种抗生素菌渣资源化处理的方法。
背景技术:
2.抗生素菌渣是抗生素生产过程中产生的固体废弃物,菌渣的主要成分为抗生素产生菌的菌丝体、未利用完的培养基、发酵过程中产生的代谢产物、培养基的降解物以及少量的抗生素等,具有含水率高、有机质含量高、蛋白含量高和纤维含量低等特点,其中粗蛋白含量达到30%~40%。抗生素菌渣还含有残留抗生素及代谢中间产物等,是一种特殊的危险废物,如处置不当,会对生态环境以及人体健康产生潜在的危害,其危害具有隐蔽性、滞后性、累计性、协同性和连带性等特点。
3.目前针对抗生素菌渣的处理方法主要为高温焚烧、堆肥和填埋等,无法从根本上克服抗生素菌渣的生物污染。对抗生素菌渣进行资源化利用,消除抗生素菌渣潜在风险,具有重要的经济和社会效益。
4.中国专利申请cn 105685105a公开了一种利用抗生素菌渣制备高蛋白菌粉的方法,但该方法制备高蛋白菌粉工艺复杂,抗生素菌渣利用量小,生产周期长,制备高蛋白菌粉时需经105℃烘干,易造成蛋白质变性,不利于大规模工业生产;中国专利申请cn 108455599a公开了一种抗生素菌渣干粉制备富含微孔的高性能活性炭的方法,制备的富含微孔的活性炭的产率较低。
技术实现要素:
5.有鉴于此,本发明的目的在于提供一种抗生素菌渣资源化处理的方法,可以规模化制备无抗高蛋白粉和碳基吸附材料,无抗高蛋白粉的蛋白质提取率高,碳基吸附材料具有比表面积大、最大碘吸附值高、产率高和灰分低的特点。
6.为了实现上述发明的目的,本发明提供以下技术方案:
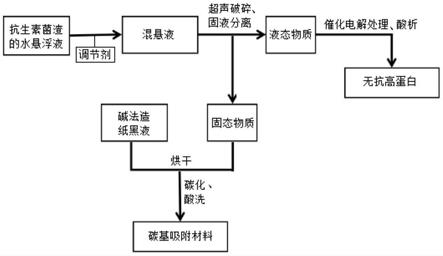
7.本发明提供了一种抗生素菌渣资源化处理的方法,包括以下步骤:
8.将抗生素菌渣的水悬浊液和调节剂混合,将所得的混悬液依次进行超声破碎和固液分离,得到液态物质和固态物质;所述调节剂为氢氧化钠和/或氢氧化钾;
9.将所述液态物质依次进行催化电解处理和酸析,得到无抗高蛋白,所述酸析的ph值为3.5~5.0;
10.将所述固态物质与碱法造纸黑液混合,烘干后依次进行碳化和酸洗,得到碳基吸附材料;所述碳化在二氧化碳或氮气条件下进行。
11.优选的,所述抗生素菌渣为螺旋霉素菌渣、红霉素菌渣、罗红霉素菌渣和盐酸林可霉素菌渣中的一种或多种。
12.优选的,所述抗生素菌渣的水悬浊液的含水率≥95%。
13.优选的,所述混悬液的ph值为12.5~13.8。
14.优选的,所述超声破碎的声能密度为5~7.5w/ml,超声破碎的时间为20~30min。
15.优选的,所述催化电解处理中的催化电极为钌铱钛电极或锡锑铜电极。
16.优选的,所述催化电解处理的电流密度为300~500ma/cm2,时间为0.5~1h。
17.优选的,所述碱法造纸黑液的固含量≥50%;所述固态物质与碱法造纸黑液的质量比为1:(3~5)。
18.优选的,所述碱法造纸黑液的ph值≥13。
19.优选的,所述碳化的温度为550~650℃,时间为30~60min。
20.本发明提供了一种抗生素菌渣资源化处理的方法,包括以下步骤:将抗生素菌渣的水悬浊液和调节剂混合,将所得的混悬液依次进行超声破碎和固液分离,得到液态物质和固态物质;所述调节剂为氢氧化钠和/或氢氧化钾;将所述液态物质依次进行催化电解处理和酸析,得到无抗高蛋白,所述酸析的ph值为3.5~5.0;将所述固态物质与碱法造纸黑液混合,烘干后依次进行碳化和酸洗,得到碳基吸附材料;所述碳化在二氧化碳或氮气条件下进行。在本发明中,抗生素菌渣的水悬浊液和调节剂混合,有利于中和抗生素菌渣的蛋白质表面的正电荷,增加蛋白质之间的静电斥力,提高蛋白质的水溶性。超声破碎能够影响抗生素菌渣中蛋白质内部分子的三维结构及活性基团分布,从而提高蛋白质与水的亲和力,增强蛋白质的溶解度,超声破碎还能够产生强烈微扰、湍动和空化效应,增强蛋白质分子之间的能量传递,使蛋白质分子更好的释放出来;抗生素菌渣中蛋白质的提取消除了固态物质在碳化过程中容易塌陷变形而使孔隙结构减少、影响碳基吸附材料品质的问题。在本发明中,催化电解处理有利于将液态物质中的抗生素逐渐分解成小分子物质,其原有的分子结构被破坏,使其失去抗菌活性。在本发明中,控制酸析的ph值为3.5~5.0,使蛋白质分子处于等电点状态,蛋白质分子表面的双电层和水化膜遭到削弱或破坏,蛋白质分子间的静电斥力基本消失,吸引力增大,增加了蛋白质分子间相互碰撞的几率,有利于降低蛋白质分子溶解度,使蛋白质最大程度发生凝聚而沉淀析出。在本发明中,所述固态物质与碱法造纸黑液的混合,碱法造纸黑液可以活化固态物质,同时碱法造纸黑液又提供碳源,提高了碳基吸附材料的碳含量;在二氧化碳或氮气条件下进行碳化,防止有机质在碳化过程中彻底分解成气体而使生成物的灰分过高,同时提高了碳基吸附材料的比表面积。本发明提供的方法简单易行,适于工业规模化生产。
21.进一步的,本发明中调节剂具有ph值调整作用,混悬液的ph值为12.5~13.8时,抗生素菌渣中蛋白质溶解率最高,提高抗生素菌渣中蛋白质提取率。
22.实施例测试结果表明,由本发明提供的方法得到的无抗高蛋白提取率高,碳基吸附材料具有比表面积大、最大碘吸附值高、产率高和灰分低的特点。
附图说明
23.图1为本发明提供的抗生素菌渣资源化处理的方法流程图;
24.图2为实施例1所得碳基吸附材料截面的sem图;
25.图3为对比例2所得碳基吸附材料截面的sem图。
具体实施方式
26.本发明提供了一种抗生素菌渣资源化处理的方法,包括以下步骤:
27.将抗生素菌渣的水悬浊液和调节剂混合,将所得的混悬液依次进行超声破碎和固液分离,得到液态物质和固态物质;所述调节剂为氢氧化钠和/或氢氧化钾;
28.将所述液态物质依次进行催化电解处理和酸析,得到无抗高蛋白,所述酸析的ph值为3.5~5.0;
29.将所述固态物质与碱法造纸黑液混合,烘干后依次进行碳化和酸洗,得到碳基吸附材料;所述碳化在二氧化碳或氮气条件下进行。
30.图1为本发明提供的抗生素菌渣资源化处理的方法流程图,下面结合图1对本发明提供的抗生素菌渣资源化处理的方法进行详细说明。
31.在本发明中,若无特殊说明,所述各组分均为本领域技术人员熟知的市售商品。
32.本发明将抗生素菌渣的水悬浊液和调节剂混合,将所得的混悬液依次进行超声破碎和固液分离,得到液态物质和固态物质。
33.在本发明中,所述抗生素菌渣优选为螺旋霉素菌渣、红霉素菌渣、罗红霉素菌渣和盐酸林可霉素菌渣中的一种或多种。本发明对所述抗生素菌渣的来源没有特殊限定,采用本领域技术人员熟知的来源即可。
34.在本发明中,所述抗生素菌渣的水悬浊液的含水率优选≥95%,更优选为95~98%。
35.在本发明中,所述抗生素菌渣的水悬浊液的制备方法优选包括:将抗生素菌渣干燥至恒重,计算出抗生素菌渣的含水率;将干燥至恒重的抗生素菌渣与水混合,得到所述抗生素菌渣的水悬浊液。
36.在本发明中,所述干燥的温度优选为100~110℃,更优选为103~108℃,最优选为105℃。
37.在本发明中,所述调节剂为氢氧化钠和/或氢氧化钾。在本发明中,所述调节剂优选以调节剂水溶液的形式使用。在本发明中,所述调节剂水溶液的浓度优选为2~4mol/l。在本发明中,所述调节剂具有调节ph值的作用,同时可以对后续碳化起到活化作用。
38.在本发明中,所述混悬液的ph值优选为12.5~13.8,更优选为12.6~13.7。在本发明中,所述调节剂的用量以保证混悬液的ph值为12.5~13.8为准。
39.在本发明中,所述超声破碎的声能密度优选为5~7.5w/ml,更优选为5.2~7.3w/ml;超声破碎的时间优选为20~30min,更优选为22~28min。在本发明中,所述超声破碎的设备优选为超声波细胞破碎机。
40.在本发明中,所述固液分离优选为离心;本发明对所述离心没有特殊限定,采用本领域技术人员熟知的离心即可。在本发明中,所述离心的转速优选为8000~10000rpm,更优选为8000~9500rpm;时间优选为5~10min,更优选为7~10min。
41.离心后,本发明得到液态物质和固态物质。本发明优选将所述固态物质干燥;本发明对所述干燥没有特殊限定,采用本领域技术人员熟知的干燥即可。
42.得到液态物质后,本发明将所述液态物质依次进行催化电解处理和酸析,得到无抗高蛋白,所述酸析的ph值为3.5~5.0。
43.在本发明中,所述催化电解处理中的催化电极优选为钌铱钛电极或锡锑铜电极。在本发明中,所述催化电解处理的设备优选为催化电解槽。在本发明中,所述催化电解处理的电流密度优选为300~500ma/cm2,更优选为320~500ma/cm2;时间优选为0.5~1h,更优选
为0.6~1h。
44.催化电解处理后,本发明优选将所得催化电解体系进行抽滤,得到上清液。
45.得到上清液后,本发明对所述上清液进行酸析,得到无抗高蛋白。
46.在本发明中,所述酸析优选为:将上清液和酸液混合,将所得酸析体系静置,使上清液中的蛋白质析出。
47.在本发明中,所述酸液优选为盐酸;所述盐酸的浓度优选为1~2mol/l,更优选为1~1.8mol/l。在本发明中,所述酸析的ph值为3.5~5.0,优选为3.7~4.8,更优选为3.7~4.6。在本发明中,所述酸液的用量以保证酸析的ph值为3.5~5.0为准。
48.在本发明中,所述上清液与酸液的混合优选为搅拌。在本发明中,所述搅拌的时间优选为10~30min,更优选为10~25min,再优选为10~20min;本发明对所述搅拌的速率没有特殊限定,采用本领域技术人员熟知的搅拌速率即可。
49.在本发明中,所述静置的时间优选为1~2h,更优选为1.5~2h。
50.酸析后,本发明优选还包括:将所得的固液混合料进行固液分离,将所得的固体进行干燥,得到无抗高蛋白。
51.在本发明中,所述固液分离优选为离心;所述离心的转速优选为8000~10000rpm,更优选为8000~9500rpm;时间优选为5~10min,更优选为7~10min。
52.在本发明中,所述干燥优选为冷冻干燥。在本发明中,所述冷冻干燥的设备优选为冷冻干燥机。在本发明中,所述冷冻干燥优选为冷冻干燥至恒重。
53.冷冻干燥后,本发明所述无抗高蛋白的形态优选为无抗高蛋白粉。
54.得到固态物质后,本发明将所述固态物质与碱法造纸黑液混合,烘干后依次进行碳化和酸洗,得到碳基吸附材料。
55.在本发明中,所述碱法造纸黑液的固含量优选≥50%,更优选为50~75%。本发明对所述碱法造纸黑液的来源没有特殊限定,采用本领域技术人员熟知的来源即可。在本发明中,所述碱法造纸黑液的成分优选包括:有机质、钙、铝、锰、硅、氮、磷和钾。在本发明中,所述碱法造纸黑液的ph值优选≥13。
56.在本发明中,所述固态物质与碱法造纸黑液的质量比优选为1:(3~5),更优选为1:(3.2~4.8),更优选为1:(3.5~4.5)。在本发明中,所述固态物质与碱法造纸黑液的混合优选为静置混合;所述静置混合的时间优选为18~24h。
57.在本发明中,所述烘干的温度优选为105℃;时间优选为3~5h,更优选为3.5~5h。
58.在本发明中,所述碳化的温度优选为550~650℃,更优选为560~640℃,再优选为570~630℃;时间优选为30~60min,更优选为35~60min,再优选为40~55min。在本发明中,所述碳化的温度优选由室温升温得到;所述升温的速率优选为2~5℃/min,更优选为3~5℃/min。在本发明中,所述碳化在二氧化碳或氮气条件下进行。在本发明中,所述碳化的设备优选为管式炉。
59.碳化后,本发明优选将碳化产物降至室温,然后进行破碎和过筛。本发明对所述破碎没有特殊限定,采用本领域技术人员熟知的破碎即可。在本发明中,所述过筛用筛网的目数优选为80目。本发明取筛下物进行后续酸洗。
60.在本发明中,所述酸洗优选为:将所述碳化产物与酸洗用酸混合,搅拌。
61.在本发明中,所述酸洗用酸优选为盐酸;所述盐酸的浓度优选为0.5~1mol/l,更
优选为0.5~0.9mol/l。在本发明中,所述碳化产物与酸洗用酸的质量比优选为1:(5~8),更优选为1:(5~7),最优选为1:5。在本发明中,所述搅拌的时间优选为30~60min,更优选为30~50min;本发明对所述搅拌的速率没有特殊限定,采用本领域技术人员熟知的速率即可。在本发明中,所述酸洗有利于去除碳化产物附着的碱金属氧化物,同时有利于降低碳基吸附材料的灰分。
62.酸洗后,本发明优选还包括:将酸洗所得酸洗物料依次进行水洗和干燥,得到碳基吸附材料。
63.本发明对所述水洗没有特殊限定,采用本领域技术人员熟知的水洗即可。本发明优选通过水洗将酸洗物料洗涤至中性。在本发明中,所述水洗使用的水优选为蒸馏水。在本发明中,所述干燥的温度优选为100~110℃;本发明优选将水洗所得物料干燥至恒重。在本发明中,所述干燥的设备优选为干燥箱。
64.为了进一步说明本发明,下面结合实施例对本发明提供的一种抗生素菌渣资源化处理的方法进行详细地描述,但不能将它们理解为对本发明保护范围的限定。显然,所描述的实施例仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
65.实施例1
66.将螺旋霉素菌渣与水混合,得到含水率≥95%的螺旋霉素菌渣的水悬浊液,将所得螺旋霉素菌渣的水悬浊液和4mol/l的氢氧化钠溶液混合,得到ph值为13.5的混悬液;
67.将所得的混悬液在声能密度为7.5w/ml条件下进行超声破碎20min,然后以8000rpm转速离心10min,得到液态物质和固态物质;
68.将所述液态物质置于含有钌铱钛电极的催化电解槽中,在400ma/cm2的电流密度条件下进行催化电解处理1h,抽滤后,将所得的上清液和1mol/l的盐酸混合至ph值为3.5,搅拌10min后静置2h进行酸析,将所得固液混合料在8000rpm的转速下离心10min,将所得的固体在冷冻干燥机中冷冻干燥至恒重,得到无抗高蛋白;
69.将所述固态物质干燥至恒重后,与固含量≥50%的碱法造纸黑液按照1:5的质量比混合,静置24h后于105℃烘箱中干燥5h,破碎后置于管式炉中,向管式炉腔体内通入二氧化碳后,以5℃/min的速率升温至550℃,于550℃保温60min进行碳化,碳化完成后自然冷却至室温,破碎并过80目筛,取筛下物为碳化产物;
70.将碳化产物与0.5mol/l的盐酸按照1:5的质量比混合,搅拌30min后抽滤,得到酸洗物料;用蒸馏水将所得酸洗物料水洗至中性,然后于烘箱中干燥至恒重,得到碳基吸附材料。
71.测试方法:菌渣固体及无抗高蛋白粉的蛋白质含量测定参照gb/t6432
‑
2018饲料中粗蛋白的测定凯氏定氮法;溶液中蛋白质含量的测定参照sn/t 2497.20
‑
2010进出口安全化学品安全试验方法第20部分bradford法测定;螺旋霉素的检测方法为液相色谱
‑
串联质谱法测定;碳基吸附材料的产率为酸洗后得到的碳基吸附材料与碳化时所用原料质量的比值;灰分参照gb/t 12496.3
‑
1999木质活性炭试验方法灰分含量的测定;碘吸附值参照gb/t 12496.8
‑
2015木质活性炭试验方法碘吸附值的测定;比表面积利用比表面积测定仪进行测定。
72.经测试,超声破碎所得液态物质中蛋白质质量为螺旋霉素菌渣中蛋白质质量的86.51%;催化电解处理后所得的上清液中未检测到螺旋霉素;催化电解处理后所得上清液中蛋白质质量为螺旋霉素菌渣中蛋白质质量的83.74%;无抗高蛋白中蛋白质含量为87.01wt.%;无抗高蛋白的质量为催化电解处理后所得上清液中蛋白质质量的97.35%;
73.碳基吸附材料的产率为34.75%,灰分为11.08%,碘吸附值为962.18mg/g,比表面积为905.71m2/g。
74.对实施例1所得碳基吸附材料的截面进行电镜扫描测试,所得sem图见图2。由图2可见,所得碳基吸附材料孔道结构丰富、均匀且呈蜂窝状。
75.实施例2
76.将螺旋霉素菌渣与水混合,得到含水率≥97%的螺旋霉素菌渣的水悬浊液,将所得螺旋霉素菌渣的水悬浊液和2mol/l的氢氧化钠溶液混合,得到ph值为13.8的混悬液;
77.将所得的混悬液在声能密度为5w/ml条件下进行超声破碎30min,然后以8000rpm转速离心10min,得到液态物质和固态物质;
78.将所述液态物质置于含有钌铱钛电极的催化电解槽中,在300ma/cm2的电流密度条件下进行催化电解处理1h,抽滤后,将所得的上清液和1mol/l的盐酸混合至ph值为5,搅拌10min后静置1.5h进行酸析,将所得固液混合料在8000rpm的转速下离心10min,将所得的固体在冷冻干燥机中冷冻干燥至恒重,得到无抗高蛋白;
79.将所述固态物质干燥至恒重后,与固含量≥50%的碱法造纸黑液按照1:4的质量比混合,静置18h后于105℃烘箱中干燥3.5h,破碎后置于管式炉中,向管式炉腔体内通入二氧化碳后,以5℃/min的速率升温至600℃,于600℃保温45min进行碳化,碳化完成后自然冷却至室温,破碎并过80目筛,取筛下物为碳化产物;
80.将碳化产物与0.5mol/l的盐酸按照1:5的质量比混合,搅拌30min后抽滤,得到酸洗物料;用蒸馏水将所得酸洗物料水洗至中性,然后于烘箱中干燥至恒重,得到碳基吸附材料。
81.经测试,超声破碎所得液态物质中蛋白质质量为螺旋霉素菌渣中蛋白质质量的78.82%;催化电解处理后所得的上清液中未检测到螺旋霉素;催化电解处理后所得上清液中蛋白质质量为螺旋霉素菌渣中蛋白质质量的77.11%;无抗高蛋白中蛋白质含量为85.13wt.%;无抗高蛋白的质量为催化电解处理后所得上清液中蛋白质质量的90.83%;
82.碳基吸附材料的产率为32.59%,灰分为13.42%,碘吸附值为795.96mg/g,比表面积为792.14m2/g。
83.实施例3
84.将螺旋霉素菌渣与水混合,得到含水率≥97%的螺旋霉素菌渣的水悬浊液,将所得螺旋霉素菌渣的水悬浊液和2mol/l的氢氧化钠溶液混合,得到ph值为12.5的混悬液;
85.将所得的混悬液在声能密度为7.5w/ml条件下进行超声破碎30min,然后以8000rpm转速离心10min,得到液态物质和固态物质;
86.将所述液态物质置于含有钌铱钛电极的催化电解槽中,在500ma/cm2的电流密度条件下进行催化电解处理0.5h,抽滤后,将所得的上清液和1mol/l的盐酸混合至ph值为5,搅拌10min后静置2h进行酸析,将所得固液混合料在8000rpm的转速下离心10min,将所得的固体在冷冻干燥机中冷冻干燥至恒重,得到无抗高蛋白;
87.将所述固态物质干燥至恒重后,与固含量≥50%的碱法造纸黑液按照1:3的质量比混合,静置24h后于105℃烘箱中干燥3h,破碎后置于管式炉中,向管式炉腔体内通入二氧化碳后,以5℃/min的速率升温至650℃,于650℃保温30min进行碳化,碳化完成后自然冷却至室温,破碎并过80目筛,取筛下物为碳化产物;
88.将碳化产物与0.5mol/l的盐酸按照1:5的质量比混合,搅拌30min后抽滤,得到酸洗物料;用蒸馏水将所得酸洗物料水洗至中性,然后于烘箱中干燥至恒重,得到碳基吸附材料。
89.经测试,超声破碎所得液态物质中蛋白质质量为螺旋霉素菌渣中蛋白质质量的82.30%;催化电解处理后所得的上清液中未检测到螺旋霉素;催化电解处理后所得上清液中蛋白质质量为螺旋霉素菌渣中蛋白质质量的81.94%;无抗高蛋白中蛋白质含量为86.27wt.%;无抗高蛋白的质量为催化电解处理后所得上清液中蛋白质质量的96.32%;
90.碳基吸附材料的产率为32.79%,灰分为12.95%,碘吸附值为927.43mg/g,比表面积为818.34m2/g。
91.对比例1
92.将螺旋霉素菌渣与水混合,得到含水率为90%的螺旋霉素菌渣的水悬浊液,将所得螺旋霉素菌渣的水悬浊液和2mol/l的氢氧化钠溶液混合,得到ph值为12.0的混悬液;
93.将所得的混悬液在声能密度为4.5w/ml条件下进行超声破碎20min,然后以8000rpm转速离心10min,得到液态物质和固态物质;
94.将所述液态物质置于含有钌铱钛电极的催化电解槽中,在200ma/cm2的电流密度条件下进行催化电解处理1h,抽滤后,将所得的上清液和1mol/l的盐酸混合至ph值为5.2,搅拌10min后静置2h进行酸析,将所得固液混合料在8000rpm的转速下离心10min,将所得的固体在冷冻干燥机中冷冻干燥至恒重,得到无抗高蛋白;
95.将所述固态物质干燥至恒重后,与固含量≥50%的碱法造纸黑液按照1:2的质量比混合,静置24h后于105℃烘箱中干燥3h,破碎后置于管式炉中,向管式炉腔体内通入二氧化碳后,以5℃/min的速率升温至600℃,于600℃保温60min进行碳化,碳化完成后自然冷却至室温,破碎并过80目筛,取筛下物为碳化产物;
96.将碳化产物与0.5mol/l的盐酸按照1:5的质量比混合,搅拌30min后抽滤,得到酸洗物料;用蒸馏水将所得酸洗物料水洗至中性,然后于烘箱中干燥至恒重,得到碳基吸附材料。
97.经测试,超声破碎所得液态物质中蛋白质质量为螺旋霉素菌渣中蛋白质质量的73.63%;催化电解处理后所得的上清液中检测到螺旋霉素含量为0.26μg/g;催化电解处理后所得上清液中蛋白质质量为螺旋霉素菌渣中蛋白质质量的71.84%;无抗高蛋白中蛋白质含量为80.06wt.%;无抗高蛋白的质量为催化电解处理后所得上清液中蛋白质质量的82.17%;
98.碳基吸附材料的产率为35.02%,灰分为13.83%,碘吸附值为619.74mg/g,比表面积为206.62m2/g。
99.对比例2
100.将螺旋霉素菌渣在105℃烘箱中干燥至恒重,得到干菌渣;将所得干菌渣与固含量≥50%的碱法造纸黑液按照1:5的质量比混合,静置24h后于105℃烘箱中干燥3h,破碎后置
于管式炉中,向管式炉腔体内通入二氧化碳后,以5℃/min的速率升温至550℃,于550℃保温60min进行碳化,碳化完成后自然冷却至室温,破碎并过80目筛,取筛下物为碳化产物;
101.将碳化产物与0.5mol/l的盐酸按照1:5的质量比混合,搅拌30min后抽滤,得到酸洗物料;用蒸馏水将所得酸洗物料水洗至中性,然后于烘箱中干燥至恒重,得到碳基吸附材料。
102.经测试,碳基吸附材料的产率为32.36%,灰分为15.76%,碘吸附值为562.71mg/g,比表面积为192.04m2/g。
103.对对比例2所得碳基吸附材料的截面进行电镜扫描测试,所得sem图见图3。由图3可见,所得碳基吸附材料孔道结构较少,且多为小孔和微孔,孔道分布不均匀。
104.以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1