一种吸附剂及其制备方法和应用
1.本发明属于超分子材料技术领域,具体涉及一种吸附剂及其制备方法和应用。
背景技术:
2.篮子状构型和不同尺寸大小的杯[n]芳烃可特异性识别金属离子、染料、气体和一些有机微污染物。众所周知,作为继冠醚和环糊精之后的第三代超分子,杯[n]芳烃是一种具有内在固有孔隙的大环化合物,可以通过甲醛和对叔丁基苯酚较为容易的制备得到。
[0003]
为提高客体分子的亲和力,通过简单的化学修饰,使杯[n]芳烃在上下边缘引入活性基团,以及与具有特定结构和官能团的小分子结合制备有机多孔聚合物网络,以扩大在样品前处理介质中的应用范围。
[0004]
相关技术中,大多数研究人员将杯[n]芳烃大环主体分子与其他客体分子通过非共价相互作用形成超分子有机骨架,这种通过主客体配位作用形成一系列具有特定功能的材料,有效实现了在气体吸附,荧光传感,催化等领域的应用。然而,非共价键的动态可逆特点使超分子有机骨架富集介质缺乏足够的稳定性,使其在复杂基质样品前处理的应用中受到了一定的限制。因此,通过构筑单体衍生再聚合的方式,将经过衍生化的磺酸化杯[n]芳烃大环主体分子作为直接构筑单元,与小分子单体通过共价键形成共价交联多孔聚合物,从而制备具有超分子识别性能和开放多孔结构的新型富集介质。然而,常规的样品前处理往往需要离心、过滤等繁琐步骤。
技术实现要素:
[0005]
本发明旨在至少解决现有技术中存在的上述技术问题之一。为此,本发明提供了一种吸附剂,该吸附剂具有超顺磁性,较大的比表面积,丰富的结合位点,以及优异的稳定性和较大的吸附能力,可作为样品前处理介质应用于食品和环境分析等领域。
[0006]
本发明还提供了上述吸附剂的制备方法。
[0007]
本发明还提供了上述吸附剂的应用。
[0008]
本发明的第一方面提供了一种吸附剂,包括fe3o4内核,所述fe3o4内核外包裹有大环交联聚合物,所述fe3o4内核为氨基改性sio2颗粒包覆的超顺磁fe3o4。
[0009]
本发明的吸附剂,至少具有以下有益效果:
[0010]
本发明的吸附剂,记为mfe3o4‑
ptmc
‑
sc6a,其中,tmc指均苯三甲酰氯,sc6a指磺化杯[6]芳烃,具有超顺磁性和丰富的结合位点,作为磁固相萃取吸附剂,通过与超高效液相色谱串联质谱相结合,可用于食品和环境分析等领域的应用。
[0011]
本发明的吸附剂,是由功能化磁性fe3o4纳米颗粒为载体,杯[6]芳烃经过前修饰的磺酸化杯[6]芳烃和均苯三甲酰氯,在n,n
‑
二甲基甲酰胺溶剂中,通过亲核取代反应制备既具有杯[6]芳烃固有的大空腔结构,也具有交联的网状结构。同时,吸附剂表现出固有的超顺磁性,大的比表面积,优异的稳定性,丰富的结合位点,在目标化合物的分离富集中能表现出优异的性能。
[0012]
本发明的吸附剂,通过红外光谱、x
‑
射线衍射,振动样品磁强计,扫描电镜和透射电镜等进行表征。结果表明,吸附剂具有良好的化学稳定性,优异的吸附性能,规则疏松的球形结构,应用范围广,处理能力强。
[0013]
本发明的吸附剂,其中的磺酸化杯[6]芳烃具有主客识别相互作用,氢键相互作用,静电相互作用,π
‑
π相互作用,疏水相互作用等多种结合位点,所以对多种化合物具有较强的结合能力。能够对多种环氧衍生物具有较强的富集能力,因此在食品和环境分析等多种领域中环氧衍生物的分离、富集与检测方面具有较大的潜力。
[0014]
本发明的吸附剂,含有磺酸化杯[6]芳烃的大环空腔结构,且具有超顺磁性和内部交联的多孔网状结构。
[0015]
本发明的吸附剂,有益效果可以进一步概括为:
[0016]
本发明的吸附剂,作为样品前处理介质,具有优异的识别性能,物理化学性质稳定,制备简单,可大规模合成,通过与其他检测仪器联用,可以进一步扩大其应用范围。
[0017]
本发明的吸附剂,具有较大的比表面积,丰富的结合位点,多重作用力,吸附容量大可富集多种与多类目标化合物。
[0018]
本发明的吸附剂,具有超顺磁性,疏松的表面结构,吸附能力强,,结合动力学快,不需要额外繁琐的前处理步骤即可快速从复杂基质样品中进行分离。
[0019]
本发明的吸附剂,作为有效的吸附剂,为基于超分子大环化合物交联聚合物的制备及应用提供技术依据。
[0020]
根据本发明的一些实施方式,所述吸附剂中,均苯三甲酰氯作为fe3o4和磺酰杯芳烃的中间连接单元,fe3o4先与均苯三甲酰氯相连后,再与酰杯芳烃相连。
[0021]
根据本发明的一些实施方式,所述吸附剂中,fe3o4并未直接与磺酰杯芳烃结合,而是与小分子单体均苯三甲酰氯相连接,然后再与磺酰杯芳烃相连接,均苯三甲酰氯充当fe3o4和磺酰杯芳烃的中间连接单元。
[0022]
根据本发明的一些实施方式,具有如下结构:
[0023]
[0024]
其中,为
[0025]
为
[0026]
为fe3o4。
[0027]
本发明的第二方面提供了制备上述吸附剂的方法,所述方法为:将所述fe3o4内核与磺酸化杯[6]芳烃和均苯三甲酰氯在溶剂中进行反应。
[0028]
根据本发明的一些实施方式,所述fe3o4内核、磺酸化杯[6]芳烃和均苯三甲酰氯的质量比为(0.8~1.4):(0.8~1.4):1。
[0029]
根据本发明的一些实施方式,所述fe3o4内核、磺酸化杯[6]芳烃和均苯三甲酰氯的质量比为1.2:1.2:1。
[0030]
反应过程中,还添加有碳酸钾,均苯三甲酰氯、磺酸化杯[6]芳烃、fe3o4@sio2‑
nh2和碳酸钾的质量比为1:1.2:1.2:1。其中,碳酸钾的作用是提供碱性环境,有利于反应进行。
[0031]
根据本发明的一些实施方式,所述反应的温度为70℃~90℃。
[0032]
根据本发明的一些实施方式,所述反应的温度约为80℃。
[0033]
根据本发明的一些实施方式,所述反应的时间为36h~60h。
[0034]
根据本发明的一些实施方式,所述反应的时间约为48h。
[0035]
根据本发明的一些实施方式,所述溶剂包括n,n
‑
二甲基甲酰胺。
[0036]
反应结束后,用1mol/lhcl洗涤固体直至co2逸出停止,目的是去除未反应的碳酸钾。
[0037]
可以通过1mol/l naoh调节溶液的ph值为10,以重新获得磺酸钠。
[0038]
分别在h2o,thf和二氯甲烷中各超声洗涤三遍,目的是洗涤未反应的化合物及活化产物。
[0039]
在60℃真空干燥箱中干燥12h得到终产物吸附剂。
[0040]
根据本发明的一些实施方式,所述fe3o4内核记为fe3o4@sio2‑
nh2,制备方法为:
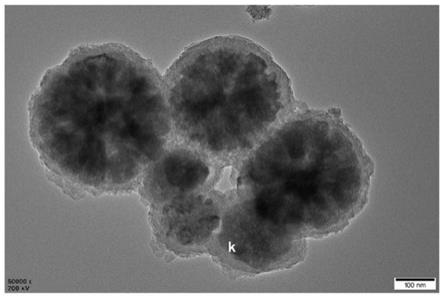
[0041]
fecl3·
6h2o,醋酸钠和聚乙二醇的质量比为1.0:2.7:0.74,所用溶剂为乙二醇,fecl3·
6h2o在乙二醇中的浓度为45g/l,剧烈搅拌15min,将粘稠溶液转移并密封在衬有特氟龙的不锈钢高压釜中。在200℃烘箱中加热12h后,随后将黑色产物在外加磁场的作用下收集并通过乙醇和超纯水各洗涤3次,在外加磁场作用下收集产物,最后将磁性fe3o4纳米颗
粒在60℃下真空干燥12h。
[0042]
由制备的磁性fe3o4纳米颗粒加入无水乙醇和去离子水体积比为4:1的溶液中,加入氢氧化铵和正硅酸乙酯体积比为5:1的混合溶剂,磁性fe3o4纳米颗粒在溶剂中的浓度为20g/l,反应时间为12h,反应温度为40℃。反应结束后,分别用去离子水洗涤三遍,在外加磁场作用下收集产物,在60℃真空干燥箱中干燥12h得到fe3o4@sio2(fe3o4@sio2表示sio2颗粒包覆的超顺磁fe3o4)备用。
[0043]
由前步骤制备的fe3o4@sio2和体积比为10:1的甲苯和(3
‑
氨基丙基)
‑
三乙氧基硅烷的混合溶剂中,fe3o4@sio2在溶剂中的浓度为27g/l,反应时为24h,反应温度为80℃,反应结束后,分别用乙醇和去离子水分别洗涤三遍,在外加磁场作用下收集产物,在60℃真空干燥箱中干燥12h得到fe3o4@sio2‑
nh2备用。
[0044]
虽然可以从市面上购买现成的磁性fe3o4纳米颗粒,但会存在分散性不够,粒径尺寸不均一,易氧化等缺点。
[0045]
虽然可以直接包裹fe3o4或者fe3o4@sio2,但会存在一些问题,如果直接包裹fe3o4会导致fe3o4易氧化,降低其磁饱和强度,影响其从复杂样品中快速分离。同时考虑在fe3o4@sio2的基础上再修饰氨基,主要是由于在最后一步的反应中(合成吸附剂的步骤中),修饰的氨基更有利于与均苯三甲酰氯反应,从而将功能化的fe3o4引入到聚合物中,并且未反应的氨基可以与目标化合物形成氢键,增加其识别位点,有利于吸附目标化合物,综上所述,本方案选择包裹fe3o4@sio2‑
nh2,而不是直接包裹fe3o4或者fe3o4@sio2。
[0046]
根据本发明的一些实施方式,所述磺酸化杯[6]芳烃的制备方法为:
[0047]4‑
叔丁基杯[6]芳烃,苯酚和alcl3的质量比为1:0.6:1,反应溶剂为甲苯,4
‑
叔丁基杯[6]芳烃在甲苯溶液中的浓度为88g/l,室温下机械搅拌反应1h,反应结束后,用甲苯和冰水体积比为1:1的冰水猝灭反应,分出有机相,通过旋转蒸发仪蒸出溶剂得到黄色残余物,使用冰水与甲醇体积比为1:0.6的甲醇洗涤,不溶物通过1:0.3的ch3oh
‑
chcl3重结晶,在60℃的真空干燥箱中干燥12h得到白色粉末固体杯[6]芳烃。
[0048]
杯[6]芳烃在浓硫酸中的浓度为33g/l,反应时间为3h,反应温度为80℃,反应结束后冷却至室温,通过玻璃漏斗过滤并将不溶物溶于水中,用baco3中和并除去baso4沉淀,通过加入na2co3调节溶液的ph值为8~9,加入乙醇,得到白色固体,在60℃真空干燥箱中干燥12h得到白色粉末磺酸化杯[6]芳烃。
[0049]
本发明的第三方面提供了上述的吸附剂在环氧衍生物吸附分离中的应用。
[0050]
本发明的第四方面提供了上述的吸附剂在食品分析与环境分析中的应用。
[0051]
本发明将磁性纳米颗粒作为载体引入到超分子交联聚合物中,使其可简单快速的从复杂基质中分离。吸附剂所提供的超顺磁性,大的比表面积,高密度的主体识别位点,多孔结构等特性为客体分子提供丰富的结合位点和多重作用力,因此在食品分析与环境分析中具有较大的应用潜力。
附图说明
[0052]
图1为本发明实施例制备的吸附剂的振动样品磁强计表征图。
[0053]
图2为本发明实施例制备的吸附剂的x射线衍射表征图。
[0054]
图3为本发明实施例制备的吸附剂的热重表征图。
[0055]
图4为本发明实施例制备的吸附剂的x射线光电子能谱表征图。
[0056]
图5为本发明实施例制备的吸附剂的扫描电镜表征图。
[0057]
图6为本发明实施例制备的吸附剂的透射电镜表征图。
[0058]
图7为本发明实施例制备的吸附剂经不同溶剂处理后的红外表征图。
[0059]
图8为本发明实施例制备的吸附剂对13种环氧衍生的吸附容量表征图。
[0060]
图9为本发明实施例制备的吸附剂对13种环氧衍生物的可重复萃取性能图。
[0061]
图10为本发明实施例制备的吸附剂对金属薄壁容器经4%(体积分数)乙酸模拟液在40℃下迁移3d,5d,10d中13种环氧衍生物迁移量的检测。
[0062]
图11为本发明实施例制备的吸附剂对金属薄壁容器经10%(体积分数)乙醇模拟液在40℃下迁移3d,5d,10d中13种环氧衍生物迁移量的检测。
[0063]
图12为本发明实施例制备的吸附剂对金属薄壁容器经20%(体积分数)乙醇模拟液在40℃下迁移3d,5d,10d中13种环氧衍生物迁移量的检测。
[0064]
图13为本发明实施例制备的吸附剂对金属薄壁容器经50%(体积分数)乙醇模拟液在40℃下迁移3d,5d,10d中13种环氧衍生物迁移量的检测。
具体实施方式
[0065]
以下是本发明的具体实施例,并结合实施例对本发明的技术方案作进一步的描述,但本发明并不限于这些实施例。
[0066]
实施例1
[0067]
本实施例具体制备了一种吸附剂,该吸附剂包括fe3o4内核,fe3o4内核外包裹有大环交联聚合物,fe3o4内核为氨基改性超顺磁fe3o4@sio2颗粒。
[0068]
本实施例制备的吸附剂,记为mfe3o4‑
ptmc
‑
sc6a,其中,tmc指均苯三甲酰氯,sc6a指磺化杯[6]芳烃
[0069]
本实施例制备的吸附剂,结构表示为:
[0070]
[0071]
其中,为
[0072]
为
[0073]
为fe3o4。
[0074]
制备方法为:将fe3o4内核与磺酸化杯[6]芳烃和均苯三甲酰氯在溶剂中进行反应。
[0075]
反应过程中,还添加有碳酸钾,均苯三甲酰氯、磺酸化杯[6]芳烃、fe3o4@sio2‑
nh2和碳酸钾的质量比为1:1.2:1.2:1。
[0076]
反应的温度为80℃。反应的时间为48h。
[0077]
溶剂为n,n
‑
二甲基甲酰胺。
[0078]
反应结束后,用1mol/lhcl洗涤固体直至co2逸出停止,通过1mol/l naoh调节溶液的ph值为10,以重新获得磺酸钠。分别在h2o,thf和二氯甲烷中各超声洗涤三遍。最后在60℃真空干燥箱中干燥12h得到终产物吸附剂。
[0079]
fe3o4内核记为fe3o4@sio2‑
nh2,指氨基改性sio2颗粒包覆的超顺磁fe3o4,制备方法为:
[0080]
fecl3·
6h2o,醋酸钠和聚乙二醇的质量比为1.0:2.7:0.74,所用溶剂为乙二醇,fecl3·
6h2o在乙二醇中的浓度为45g/l,剧烈搅拌15min,将粘稠溶液转移并密封在衬有特氟龙的不锈钢高压釜中。在200℃烘箱中加热12h后,随后将黑色产物在外加磁场的作用下收集并通过乙醇和超纯水各洗涤3次,在外加磁场作用下收集产物,最后将磁性fe3o4纳米颗粒在60℃下真空干燥12h。
[0081]
由制备的磁性fe3o4纳米颗粒加入无水乙醇和去离子水体积比为4:1的溶液中,加入氢氧化铵和正硅酸乙酯体积比为5:1的混合溶剂,磁性fe3o4纳米颗粒在溶剂中的浓度为20g/l,反应时间为12h,反应温度为40℃。反应结束后,分别用去离子水洗涤三遍,在外加磁场作用下收集产物,在60℃真空干燥箱中干燥12h得到fe3o4@sio2备用。
[0082]
由前步骤制备的fe3o4@sio2和体积比为10:1的甲苯和(3
‑
氨基丙基)
‑
三乙氧基硅烷的混合溶剂中,fe3o4@sio2在溶剂中的浓度为27g/l,反应时为24h,反应温度为80℃,反应结束后,分别用乙醇和去离子水分别洗涤三遍,在外加磁场作用下收集产物,在60℃真空干燥箱中干燥12h得到fe3o4@sio2‑
nh2备用。
[0083]
虽然可以从市面上购买现成的磁性fe3o4纳米颗粒,但会存在分散性不够,粒径尺
寸不均一,易氧化等缺点。
[0084]
虽然可以直接包裹fe3o4或者fe3o4@sio2,但会存在一些问题,如果直接包裹fe3o4会导致fe3o4易氧化,降低其磁饱和强度,影响其从复杂样品中快速分离。同时考虑在fe3o4@sio2的基础上再修饰氨基,主要是由于在最后一步的反应中(合成吸附剂的步骤中),修饰的氨基更有利于与均苯三甲酰氯反应,从而将功能化的fe3o4引入到聚合物中,并且未反应的氨基可以与目标化合物形成氢键,增加其识别位点,有利于吸附目标化合物,综上所述,本方案选择包裹fe3o4@sio2‑
nh2,而不是直接包裹fe3o4或者fe3o4@sio2。
[0085]
磺酸化杯[6]芳烃的制备方法为:
[0086]4‑
叔丁基杯[6]芳烃,苯酚和alcl3的质量比为1:0.6:1,反应溶剂为甲苯,4
‑
叔丁基杯[6]芳烃在甲苯溶液中的浓度为88g/l,室温下机械搅拌反应1h,反应结束后,用甲苯和冰水体积比为1:1的冰水猝灭反应,分出有机相,通过旋转蒸发仪蒸出溶剂得到黄色残余物,使用冰水与甲醇体积比为1:0.6的甲醇洗涤,不溶物通过1:0.3的ch3oh
‑
chcl3重结晶,在60℃的真空干燥箱中干燥12h得到白色粉末固体杯[6]芳烃。
[0087]
杯[6]芳烃在浓硫酸中的浓度为33g/l,反应时间为3h,反应温度为80℃,反应结束后冷却至室温,通过玻璃漏斗过滤并将不溶物溶于水中,用baco3中和并除去baso4沉淀,通过加入na2co3调节溶液的ph值为8~9,加入乙醇,得到白色固体,在60℃真空干燥箱中干燥12h得到白色粉末磺酸化杯[6]芳烃。
[0088]
采用振动样品磁强计对上述制备的吸附剂进行表征,其磁滞回线图谱如图1所示,从图1中可以看出,该聚合物的饱和磁化强度为40.4emu/g,无剩余磁和矫顽力,表明制备的吸附剂具有超顺磁性。
[0089]
采用x射线衍射对上述制备的吸附剂进行表征,其x射线衍射图谱如图2所示,从图2中可以看出,该聚合物有6个特征衍射峰(220
°
,311
°
,400
°
,422
°
,511
°
,440
°
),这些特征衍射峰与具有尖晶石结构的fe3o4(220),(311),(400),(422),(511)和(440)平面相匹配。结果表明,磁性fe3o4在经过修饰和包裹后并不改变其晶型结构。
[0090]
采用热重对上述制备的吸附剂进行表征,其热重图谱如图3所示,从图3中可以看出,该聚合物在400℃下相对稳定,在700℃时的重量损失大约为27.2wt%,这意味着在磁性纳米颗粒表面所包裹的聚合物壳的含量很高。
[0091]
采用x射线光电子能谱对上述制备的吸附剂进行表征,其x射线光电子能谱图谱如图4所示,从图中可以看出,该聚合物中存在c
1s
,o
1s
,n
1s
,si
2p
,fe
2p
和s
2p
峰,表明磁性fe3o4表面已成功官能化。
[0092]
采用扫描电镜对上述制备的吸附剂进行表征,其扫描电镜图谱如图5所示,从图中可以看出,该聚合物粒径分布均匀,平均粒径大约为218nm。
[0093]
采用投射电镜对上述制备的吸附剂进行表征,其投射电镜图谱如图6所示,从图中可以看出,该聚合物比表面积大且表面结构疏松,所包裹的聚合物厚度大约为38nm。
[0094]
采用红外光谱对上述制备的吸附剂(mfe3o4‑
ptmc
‑
sc6a)进行表征,其红外图谱如图7所示,从图中可以看出,该聚合物在1372cm
‑1和1619cm
‑1处的吸收峰表明形成了酯,在经过不同溶剂浸泡后与未经浸泡前相比,其特征吸收峰未发生变化,表明所制备的材料具有良好的稳定性。
[0095]
将吸附剂作为样品前处理介质,在一系列浓度条件下,通过静态平衡吸附实验评
估吸附剂对食品接触材料中13种环氧衍生物的吸附性能,如图8所示,结果表明,即使在1mg/l的混合标准浓度下,材料仍未达到吸附平衡,仍具有较大的上升空间,这与其大的比表面积和丰富的结合位点密不可分。其中,badge指双酚a
‑
二缩水甘油醚(bisphbenol a diglycidyl ether),bfdge指双酚f
‑
二缩水甘油醚(bisphbenol f diglycidyl ether),noge指酚醛甘油醚酯。
[0096]
将本实施例制备的吸附剂作为磁固相萃取吸附剂,通过与高效液相色谱串联质谱相结合应用于金属薄壁容器中13种环氧衍生物的分离、富集与检测。
[0097]
吸附剂用于13种环氧衍生物的吸附分离,如图9所示,以13种环氧衍生物(badge,badge
·
h2o,badge
·
2h2o,badge
·
hcl,badge
·
2hcl,badge
·
h2o
·
hcl,bfdge,bfdge
·
2h2o,bfdge
·
2hcl,3r
‑
noge,4r
‑
noge,5r
‑
noge,6r
‑
noge)为研究对象,通过磁固相萃取与超高效液相色谱串联质谱相结合,使用四氢呋喃和甲醇的混合溶剂超声洗脱吸附在材料上的目标化合物,来评估连续经过15次吸附
‑
解吸循环后材料的可重复使用性。具体色谱方法为:
[0098]
色谱条件:流动相为0.01mol/l甲酸铵(a)和甲醇(b)。流速为0.3ml/min。等度洗脱:0
‑
5min,10%a
‑
90%b。色谱分离的柱温为35℃,进样量为5μl。
[0099]
萃取条件:使用10ml 10μg/l的13种混合标准溶液,吸附剂的量为10mg,萃取溶剂为水,萃取时间为10min。
[0100]
洗脱条件:洗脱溶剂为四氢呋喃:甲醇=1:1(v/v),洗脱时间为5min,洗脱体积为4ml。
[0101]
所有目标物均在正电喷雾离子化(esi)模式下以多重反应监测模式(mrm)进行实验,雾化气体流速为3l/min;加热气体流速为10l/min;加热块温度为400℃;干燥气体流速为10l/min;界面电压:4.0kv;界面温度为300℃。
[0102]
连续经过15个吸附
‑
解吸循环后,材料对13种环氧衍生物的吸附性能基本上没有下降,目标化合物吸附量的rsd小于9.1,表明所制备的材料具有良好的可重复使用性和再生性能,简单的再生也将使材料成为实际应用中潜在的吸附剂。
[0103]
还将吸附剂结合超高效液相色谱串联质谱应用于金属薄壁容器中13种环氧衍生物的分离、富集与检测。
[0104]
样品前处理:分别将罐子经特定模拟液(4%乙酸、10%乙醇、20%乙醇、50%乙醇,体积分数)在40℃下迁移3d,5d,10d,迁移液的用量依据6dm2食品接触材料及制品接触1l的食品或食品模拟液的原则进行选择。迁移完成后,将10ml模拟液用于后续磁固相萃取步骤。
[0105]
色谱条件:流动相为0.01mol/l甲酸铵(a)和甲醇(b)。流速为0.3ml/min。等度洗脱:0
‑
5min,10%a
‑
90%b。色谱分离的柱温为35℃,进样量为5μl。
[0106]
萃取条件:使用10ml 10μg/l的13种混合标准溶液,吸附剂的量为10mg,萃取溶剂为水,萃取时间为10min。
[0107]
洗脱条件:洗脱溶剂为四氢呋喃:甲醇=1:1(v/v),洗脱时间为5min,洗脱体积为4ml。
[0108]
所有目标物均在正电喷雾离子化(esi)模式下以多重反应监测模式(mrm)进行实验,雾化气体流速为3l/min;加热气体流速为10l/min;加热块温度为400℃;干燥气体流速为10l/min;界面电压:4.0kv;界面温度为300℃。
[0109]
吸附剂(mfe3o4‑
ptmc
‑
sc6a)作为磁固相萃取吸附剂结合超高效液相色谱串联质谱,应用于检测金属薄壁容器经特定模拟液(4%乙酸、10%乙醇、20%乙醇、50%乙醇(体积分数))在40℃下迁移3d,5d,10d的迁移量,如图10至图13所示,在四种模拟液中都存在badge
·
h2o,badge
·
2h2o,badge
·
2hcl,badge
·
h2o
·
hcl,bfdge五种环氧衍生物,而badge仅在10%乙醇、20%乙醇和50%乙醇模拟液中存在。bfdge及其衍生物在四种模拟液中均有检测,而仅有3r
‑
noge在10%乙醇、20%乙醇和50%乙醇模拟液中存在。并且结果表明在40℃不同模拟液的迁移条件下,环氧衍生物的迁移量随时间的增加而增加,但基本上在10d内可迁移完全。
[0110]
上面结合实施例对本发明作了详细说明,但是本发明不限于上述实施例,在所属技术领域普通技术人员所具备的知识范围内,还可以在不脱离本发明宗旨的前提下作出各种变化。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1