烷基芳烃脱氢催化剂及其制备方法和应用与流程
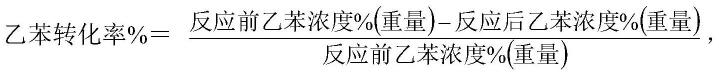
1.本发明属于催化剂制备领域,特别涉及一种用于烷基芳烃脱氢制备烷烯基芳烃的催化剂及其制备方法和应用。
背景技术:
2.工业上烷烯基芳烃主要是由烷基芳烃催化脱氢制得,该方法的关键之一是选择一种活性高、选择性高、稳定性好的脱氢催化剂。烷基芳烃催化脱氢催化剂主要分两类:一类是fe-k-cr系列,如美国专利us4152300、us4144197、中国专利cn87100517,虽然该类催化剂的活性和稳定性较好,但由于含有对环境不太友好的铬,已被淘汰;另一类是fe-k-ce-mo系列,如中国专利cn201210021958.x、cn200875018406.5,此类催化剂不仅用ce、mo替代了cr,而且催化剂的活性和稳定性较前者有较大幅度的提高,它存在的主要问题是k2o含量高,一般都在10~20%。
3.烷基芳烃脱氢催化剂普遍采用的是以氧化铁为主要活性组分、氧化钾为主要助催化剂的铁系催化剂,但钾在高温水蒸汽冲刷下缓慢流失与迁移,是催化剂不可逆失活的主要原因之一,目前降低钾含量是烷基芳烃脱氢催化剂开发的主流。
4.就工业上乙苯脱氢生产苯乙烯而言,其年产量大都在10万吨/年以上,中国已有多套50万吨/年以上规模装置,高的反应温度导致副产物多,能耗高,一直是困扰苯乙烯生产厂家的难题。脱氢反应温度如能降低5℃乃至10℃,空速如能提高,从而获得更多苯乙烯产品、降低单位产品物耗和能耗,对工业装置而言,不要改动任何设备,不需增加投资,一年就能节省水蒸汽、电方面的大量开支,另外由于脱氢反应在低温下运行,对于延长催化剂寿命、降低设备材质耐高温要求,也有积极意义。因此,开发一种适于低温、高空速条件下运行、具有更高活性和更好稳定性的低钾催化剂,大幅度降低能耗,一直是研究人员努力的方向。
技术实现要素:
5.本发明所要解决的技术问题之一是现有技术中存在的低钾含量的烷基芳烃脱氢催化剂在低温、高空速条件下活性低、稳定性差的问题,提供一种新的用于烷基芳烃脱氢制备烷烯基芳烃的催化剂。该催化剂钾含量低,低温、高空速条件下用于烷基芳烃脱氢制备烷烯基芳烃具有很好的活性和稳定性,从而获得更多产品、有效降低能耗、延长装置运行周期、减少催化剂更新费用及由于频繁更换催化剂造成的产量损失。
6.本发明所要解决的技术问题之二是提供一种与解决技术问题之一相对应的催化剂的制备方法。
7.本发明所要解决的技术问题之三是提供一种与解决技术问题之一相对应的烷基芳烃脱氢制备烷烯基芳烃的方法。
8.为解决上述技术问题之一,本发明采用的技术方案如下:
9.一种烷基芳烃脱氢制备烷烯基芳烃的催化剂,以催化剂的质量为基准,包括以下
组份:
10.(a)66%~79%的fe2o3;
11.(b)2.4%~5.2%的k2o;
12.(c)6.1%~11.9%的ceo2;
13.(d)0.5%~5%的wo3;
14.(e)0.5%~5%的sro;
15.(f)0.8%~7.7%的mnfe2o4;
16.(g)0.5%~5%的k2tio3;
17.(h)0.5%~2.5%的组分h,组分h选自geo2、sno2或pbo2的至少一种。
18.上述技术方案中,所述催化剂中,mnfe2o4含量优选为2%~6%。
19.上述技术方案中,所述催化剂中,k2tio3含量优选为1%~4%。
20.上述技术方案中,所述催化剂中优选不含有氧化钼。
21.上述技术方案中,所述催化剂中,ceo2源于草酸铈和/或氢氧化铈。
22.上述技术方案中,所述催化剂中,还可以含有其它组分,比如nb2o5。
23.上述技术方案中,所述催化剂中不含粘结剂组分。为解决上述技术问题之二,本发明采用的技术方案如下:
24.上述技术问题之一的技术方案中所述催化剂的制备方法,包括以下步骤:
25.将fe源、k源、ce源、w源、sr源、mnfe2o4、k2tio3及组分h源(组分h源选自ge源或sn源或pb源中的至少一种)和致孔剂混合成型,经干燥和焙烧,得到所述的催化剂。
26.上述技术方案中,优选地,将fe源、k源、ce源、w源、sr源、及选自ge源或sn源或pb源中的至少一种和致孔剂混合,然后加入mnfe2o4、k2tio3继续混合,再加入水(优选为脱离子水)混合成型,得到催化剂前驱体;催化剂前驱体经过干燥和焙烧,得到所述催化剂。
27.上述技术方案中,所述fe源为氧化铁红和氧化铁黄,其重量配比优选为氧化铁红:氧化铁黄=(0.5~1.2):1。
28.上述技术方案中,所述k源为碳酸钾。所述ce源为草酸铈和/或氢氧化铈;所述w源为钨盐或氧化物中的至少一种。所述sr源为氧化物或碳酸盐中的至少一种。所述ge源、sn源、pb源各自独立选自其盐或氧化物中的至少一种。
29.上述技术方案中,所述致孔剂选自石墨、聚苯乙烯微球、羧甲基纤维素钠中的至少一种,优选为羧甲基纤维素钠,其加入量为催化剂总重量的2.4%~6.1%。
30.上述技术方案中,催化剂制备过程中不加粘结剂,所述粘结剂包括高岭土、硅藻土和水泥中的一种。
31.上述技术方案中,脱离子水的加入量没有特别限制,本领域技术人员为了挤出需要可以合理掌握干湿度,例如但不限于加入量为催化剂原料总重19.8%~35.2%。
32.上述技术方案中,催化剂制备过程中,所述干燥优选采用三步法,即35~55℃干燥2~4小时,75~95℃干燥2~4小时,115~135℃干燥2~4小时。
33.上述技术方案中,催化剂制备过程中,所述焙烧优选采用三步法,即425~575℃焙烧0.5~4小时,625~775℃焙烧0.5~4小时,820~875℃焙烧0.5~4小时。
34.为解决上述技术问题之三,提供了一种烷基芳烃脱氢制备烷烯基芳烃的方法,其中采用本发明所述的催化剂。
35.上述技术方案中,所述方法包括:以烷基芳烃为原料,在本发明催化剂的存在下进行脱氢反应而获得烷烯基芳烃的步骤。
36.上述技术方案中,烷基芳烃可以为乙苯、二乙苯、甲乙苯等。所述烷烯基芳烃相应地为苯乙烯、二乙烯基苯、甲基苯乙烯等。
37.上述技术方案中,所述方法的反应条件如下:反应温度575~595℃,液体体积空速1.2~2.0小时-1
、水比(重量)1.0~2.0、压力为-70~0kpa。
38.与现有技术相比,本发明具有如下有益技术效果:
39.发明人经研究发现,通过在铁-钾-铈-钨-锶烷基芳烃脱氢催化体系中添加适量铁酸锰、适量钛酸钾以及选自geo2、sno2和pbo2的至少一种,能够明显提高低钾含量催化剂在低温、高空速条件下的活性和稳定性,其可能的原因是mnfe2o4和k2tio3的加入增进了三价铁的耐还原能力,降低了钾的不稳定性,另一方面不引入粘结剂组分,减少了低效物质的引入,进一步稳定和分散了催化剂的活性相,加快了水蒸汽与催化剂表面积炭发生水煤气反应的速率,增强了催化剂的自再生能力。
40.采用本发明催化剂在-50kpa、乙苯体积空速1.6小时-1
、反应温度585℃、水/乙苯重量比1.4条件下,乙苯转化率可以达到73.7%,在低温、高空速条件下稳定运行3000小时后乙苯转化率仍可以达到73.4%,脱氢液水相中基本不能检测到铁离子浓度,取得了较好的技术效果。
具体实施方式
41.下面通过实施例对本发明作进一步的详细阐述。
42.本发明实施例和比较例中,催化剂在等温式固定床中进行活性评价,对乙苯脱氢制苯乙烯催化剂活性评价而言,过程简述如下:
43.将脱离子水和乙苯分别经计量泵输入预热混合器,预热混合成气态后进入反应器,反应器采用电热丝加热,使之达到预定温度。反应器内径为1
″
的不锈钢管,内装填100毫升、粒径3毫米的催化剂。由反应器流出的反应物经水冷凝后用气相色谱仪分析其组成。以乙苯脱氢制备苯乙烯为例,
44.乙苯转化率、苯乙烯选择性按以下公式计算:
[0045][0046][0047]
样品的活性组分流失情况是对脱氢液水相中铁离子浓度进行测定,仪器为pe-zeeman 5000型原子吸收光谱仪,分析线404.4nm,灯电流10ma,狭缝0.7nm。
[0048]
[实施例1]
[0049]
将相当于36.74份fe2o3的氧化铁红、相当于36.74份fe2o3的氧化铁黄、相当于4.66份k2o的碳酸钾、相当于9.64份ceo2的草酸铈、相当于2.58份wo3的钨酸铵、相当于2.38份sro的碳酸锶、1.68份geo2及5.69份羧甲基纤维素钠在捏合机中搅拌0.2小时,加入3.16份mnfe2o4和2.42份k2tio3,再搅拌1.8小时,加入占催化剂原料总重27.7%的脱离子水,拌和
0.55小时,取出挤条,挤成直径3毫米、长6毫米的颗粒,放入烘箱,45℃烘2.5小时,85℃烘2.5小时,128℃烘3小时,然后置于马福炉中,于555℃焙烧2.5小时,675℃焙烧3小时,865℃焙烧3小时得到成品催化剂,催化剂组成列于表1。
[0050]
将100毫升催化剂装入反应器,在-50kpa、液体体积空速1.6小时-1
、反应温度585℃、水/乙苯重量比1.4条件下进行活性评价,评价结果列于表2。在评价3000小时,收集脱氢液,水相、油相分离后,对脱氢液水相中铁离子浓度进行测定,测试结果列于表2。
[0051]
[比较例1]
[0052]
除了不用mnfe2o4、k2tio3和geo2以外,其余组分的相对比例关系、催化剂制备方法和催化剂评价条件以及分析方法均与实施例1相同,具体组成为:相当于36.74份fe2o3的氧化铁红、相当于36.74份fe2o3的氧化铁黄、相当于4.66份k2o的碳酸钾、相当于9.64份ceo2的草酸铈、相当于2.58份wo3的钨酸铵、相当于2.38份sro的碳酸锶及5.28份羧甲基纤维素钠,催化剂组成列于表1。测试结果列于表2。
[0053]
[比较例2]
[0054]
除了用氧化铁红、氧化铁黄和mno代替同比例的mnfe2o4以外,k2o和tio2代替同比例的k2tio3外,其余组分的相对比例关系、催化剂制备方法和催化剂评价条件以及分析方法同实施例1,具体为:
[0055]
将相当于37.83份fe2o3的氧化铁红、相当于37.83份fe2o3的氧化铁黄、相当于5.96份k2o的碳酸钾、相当于9.64份ceo2的草酸铈、相当于2.58份wo3的钨酸铵、相当于2.38份sro的碳酸锶、1.68份geo2及5.69份羧甲基纤维素钠在捏合机中搅拌0.2小时,加入0.98份mno和1.12份tio2,再搅拌1.8小时,加入占催化剂原料总重27.7%的脱离子水,拌和0.55小时,取出挤条,挤成直径3毫米、长6毫米的颗粒,放入烘箱,45℃烘2.5小时,85℃烘2.5小时,128℃烘3小时,然后置于马福炉中,于555℃焙烧2.5小时,675℃焙烧3小时,865℃焙烧3小时得到成品催化剂,催化剂组成列于表1。测试结果列于表2。
[0056]
[实施例2]
[0057]
按实施例1的方法制备催化剂和测试催化剂,所不同的是用sno2替代geo2。
[0058]
催化剂的组成见表1,测试结果列于表2。
[0059]
[比较例3]
[0060]
除了用氧化铁红、氧化铁黄和mno代替同比例的mnfe2o4以外,k2o和tio2代替同比例的k2tio3外,其余组分的相对比例关系、催化剂制备方法和催化剂评价条件以及分析方法同实施例2,具体为:
[0061]
将相当于37.83份fe2o3的氧化铁红、相当于37.83份fe2o3的氧化铁黄、相当于5.96份k2o的碳酸钾、相当于9.64份ceo2的草酸铈、相当于2.58份wo3的钨酸铵、相当于2.38份sro的碳酸锶、1.68份sno2及5.69份羧甲基纤维素钠在捏合机中搅拌0.2小时,加入0.98份mno和1.12份tio2,再搅拌1.8小时,加入占催化剂原料总重27.7%的脱离子水,拌和0.55小时,取出挤条,挤成直径3毫米、长6毫米的颗粒,放入烘箱,45℃烘2.5小时,85℃烘2.5小时,128℃烘3小时,然后置于马福炉中,于555℃焙烧2.5小时,675℃焙烧3小时,865℃焙烧3小时得到成品催化剂,催化剂组成列于表1。测试结果列于表2。
[0062]
[比较例4]
[0063]
按实施例2的方法制备催化剂和测试催化剂,所不同的是用氧化铈替代草酸铈。
[0064]
催化剂的组成见表1,测试结果列于表2。
[0065]
[比较例5]
[0066]
按实施例2的方法测试催化剂,制备方法和具体组成为:将相当于36.74份fe2o3的氧化铁红、相当于36.74份fe2o3的氧化铁黄、相当于4.66份k2o的碳酸钾、相当于9.64份ceo2的草酸铈、相当于2.58份wo3的钨酸铵、相当于2.38份sro的碳酸锶、3.16份mnfe2o4和2.42份k2tio3、1.68份sno2及5.69份羧甲基纤维素钠在捏合机中搅拌2小时,加入占催化剂原料总重27.7%的脱离子水,拌和0.55小时,取出挤条,挤成直径3毫米、长6毫米的颗粒,放入烘箱,45℃烘2.5小时,85℃烘2.5小时,128℃烘3小时,然后置于马福炉中,于555℃焙烧2.5小时,675℃焙烧3小时,865℃焙烧3小时得到成品催化剂,催化剂组成列于表1,测试结果列于表2。
[0067]
[实施例3]
[0068]
将相当于24.25份fe2o3的氧化铁红、相当于46.08份fe2o3的氧化铁黄、相当于2.35份k2o的碳酸钾、相当于11.9份ceo2的氢氧化铈、相当于4.16份wo3的钨酸铵、相当于3.35份sro的碳酸锶、0.85份pbo2、0.36份nb2o5及5.69份石墨在捏合机中搅拌0.2小时,加入5.5份mnfe2o4和1.2份k2tio3,再搅拌1.8小时,加入占催化剂原料总重27.7%的脱离子水,拌和0.55小时,取出挤条,挤成直径3毫米、长6毫米的颗粒,放入烘箱,45℃烘2.5小时,85℃烘2.5小时,128℃烘3小时,然后置于马福炉中,于555℃焙烧2.5小时,675℃焙烧3小时,865℃焙烧3小时得到成品催化剂,催化剂组成列于表1。
[0069]
按实施例1的方法评价和分析催化剂,测试结果列于表2。
[0070]
[实施例4]
[0071]
将相当于30.73份fe2o3的氧化铁红、相当于37.45份fe2o3的氧化铁黄、相当于3.95份k2o的碳酸钾、相当于6.55份ceo2的草酸铈、相当于1.72份wo3的钨酸铵、相当于4.95份sro的碳酸锶、2.15份geo2及5.69份石墨在捏合机中搅拌0.2小时,加入7.65份mnfe2o4和4.85份k2tio3,再搅拌1.8小时,加入占催化剂原料总重27.7%的脱离子水,拌和0.55小时,取出挤条,挤成直径3毫米、长6毫米的颗粒,放入烘箱,45℃烘2.5小时,85℃烘2.5小时,128℃烘3小时,然后置于马福炉中,于555℃焙烧2.5小时,675℃焙烧3小时,865℃焙烧3小时得到成品催化剂,催化剂组成列于表1。
[0072]
按实施例1的方法评价和分析催化剂,测试结果列于表2。
[0073]
[实施例5]
[0074]
将相当于35.48份fe2o3的氧化铁红、相当于38.45份fe2o3的氧化铁黄、相当于3.71份k2o的碳酸钾、相当于9.46份ceo2的草酸铈、相当于4.82份wo3的钨酸铵、相当于0.83份sro的碳酸锶、2.45份geo2及5.69份石墨在捏合机中搅拌0.2小时,加入0.95份mnfe2o4和3.85份k2tio3,再搅拌1.8小时,加入占催化剂原料总重27.7%的脱离子水,拌和0.55小时,取出挤条,挤成直径3毫米、长6毫米的颗粒,放入烘箱,45℃烘2.5小时,85℃烘2.5小时,128℃烘3小时,然后置于马福炉中,于555℃焙烧2.5小时,675℃焙烧3小时,865℃焙烧3小时得到成品催化剂,催化剂组成列于表1。
[0075]
按实施例1的方法评价和分析催化剂,测试结果列于表2。
[0076]
[实施例6]
[0077]
将相当于41.69份fe2o3的氧化铁红、相当于36.36份fe2o3的氧化铁黄、相当于5.05
份k2o的碳酸钾、相当于7.15份ceo2的草酸铈、相当于0.55份wo3的钨酸铵、相当于0.55份sro的碳酸锶、2.05份geo2及5.69份羧甲基纤维素钠在捏合机中搅拌0.2小时,加入1.85份mnfe2o4和4.75份k2tio3,再搅拌1.8小时,加入占催化剂原料总重27.7%的脱离子水,拌和0.55小时,取出挤条,挤成直径3毫米、长6毫米的颗粒,放入烘箱,45℃烘2.5小时,85℃烘2.5小时,128℃烘3小时,然后置于马福炉中,于555℃焙烧2.5小时,675℃焙烧3小时,865℃焙烧3小时得到成品催化剂,催化剂组成列于表1。
[0078]
按实施例1的方法评价和分析催化剂,测试结果列于表2。
[0079]
[实施例7]
[0080]
将相当于38.7份fe2o3的氧化铁红、相当于36.4份fe2o3的氧化铁黄、相当于2.35份k2o的碳酸钾、相当于8.04份ceo2的草酸铈、相当于3.41份wo3的钨酸铵、相当于1.35份sro的碳酸锶、2.35份sno2及5.69份石墨在捏合机中搅拌0.2小时,加入6.65份mnfe2o4和0.75份k2tio3,再搅拌1.8小时,加入占催化剂原料总重27.7%的脱离子水,拌和0.55小时,取出挤条,挤成直径3毫米、长6毫米的颗粒,放入烘箱,45℃烘2.5小时,85℃烘2.5小时,128℃烘3小时,然后置于马福炉中,于555℃焙烧2.5小时,675℃焙烧3小时,865℃焙烧3小时得到成品催化剂,催化剂组成列于表1。
[0081]
按实施例1的方法评价和分析催化剂,测试结果列于表2。
[0082]
[比较例6]
[0083]
将相当于42.9份fe2o3的氧化铁红、相当于26.8份fe2o3的氧化铁黄、相当于5.8份k2o的碳酸钾、相当于9.1份ceo2的草酸铈、相当于2.5份wo3的钨酸铵、相当于4.75份sro的碳酸锶、0.65份geo2及5.69份羧甲基纤维素钠在捏合机中搅拌0.2小时,加入2.6份mnfe2o4和4.9份k2tio3,再搅拌1.8小时,加入占催化剂原料总重27.7%的脱离子水,拌和0.55小时,取出挤条,挤成直径3毫米、长6毫米的颗粒,放入烘箱,45℃烘2.5小时,85℃烘2.5小时,128℃烘3小时,然后置于马福炉中,于555℃焙烧2.5小时,675℃焙烧3小时,865℃焙烧3小时得到成品催化剂,催化剂组成列于表1。
[0084]
按实施例1的方法评价和分析催化剂,测试结果列于表2。
[0085]
[比较例7]
[0086]
将相当于36.5份fe2o3的氧化铁红、相当于38.18份fe2o3的氧化铁黄、相当于5.55份k2o的碳酸钾、相当于7.75份ceo2的草酸铈、相当于2.58份wo3的钨酸铵、相当于1.38份sro的碳酸锶、1.68份geo2、1.21份水泥及5.69份羧甲基纤维素钠在捏合机中搅拌0.2小时,加入3.16份mnfe2o4和2.01份k2tio3,再搅拌1.8小时,加入占催化剂原料总重27.7%的脱离子水,拌和0.55小时,取出挤条,挤成直径3毫米、长6毫米的颗粒,放入烘箱,45℃烘2.5小时,85℃烘2.5小时,128℃烘3小时,然后置于马福炉中,于555℃焙烧2.5小时,675℃焙烧3小时,865℃焙烧3小时得到成品催化剂,催化剂组成列于表1。
[0087]
按实施例1的方法评价和分析催化剂,测试结果列于表2。
[0088]
[比较例8]
[0089]
将相当于38.36份fe2o3的氧化铁红、相当于37.42份fe2o3的氧化铁黄、相当于6.45份k2o的碳酸钾、相当于6.15份ceo2的草酸铈、相当于1.76份wo3的钨酸铵、相当于2.73份sro的碳酸锶、2.18份geo2、相当于1.55份moo3的钼酸铵及5.69份羧甲基纤维素钠在捏合机中搅拌0.2小时,加入1.62份mnfe2o4和1.78份k2tio3,再搅拌1.8小时,加入占催化剂原料总重
27.7%的脱离子水,拌和0.55小时,取出挤条,挤成直径3毫米、长6毫米的颗粒,放入烘箱,45℃烘2.5小时,85℃烘2.5小时,128℃烘3小时,然后置于马福炉中,于555℃焙烧2.5小时,675℃焙烧3小时,865℃焙烧3小时得到成品催化剂,催化剂组成列于表1。
[0090]
按实施例1的方法评价和分析催化剂,测试结果列于表2。
[0091]
[比较例9]
[0092]
将相当于36.5份fe2o3的氧化铁红、相当于37.4份fe2o3的氧化铁黄、相当于3.55份k2o的碳酸钾、相当于7.75份ceo2的草酸铈、相当于2.58份wo3的钨酸铵、相当于1.38份sro的碳酸锶、1.15份geo2及5.69份石墨在捏合机中搅拌0.2小时,加入8.15份mnfe2o4和1.54份k2tio3,再搅拌1.8小时,加入占催化剂原料总重27.7%的脱离子水,拌和0.55小时,取出挤条,挤成直径3毫米、长6毫米的颗粒,放入烘箱,45℃烘2.5小时,85℃烘2.5小时,128℃烘3小时,然后置于马福炉中,于555℃焙烧2.5小时,675℃焙烧3小时,865℃焙烧3小时得到成品催化剂,催化剂组成列于表1。
[0093]
按实施例1的方法评价和分析催化剂,测试结果列于表2。
[0094]
[比较例10]
[0095]
将29.97份fe2o3的氧化铁红、相当于44.05份fe2o3的氧化铁黄、相当于4.85份k2o的碳酸钾、相当于7.11份ceo2的草酸铈、相当于3.42份wo3的钨酸铵、相当于2.35份sro的碳酸锶、0.85份geo2及5.69份石墨在捏合机中搅拌0.2小时,加入1.65份mnfe2o4和5.75份k2tio3,再搅拌1.8小时,加入占催化剂原料总重27.7%的脱离子水,拌和0.55小时,取出挤条,挤成直径3毫米、长6毫米的颗粒,放入烘箱,45℃烘2.5小时,85℃烘2.5小时,128℃烘3小时,然后置于马福炉中,于555℃焙烧2.5小时,675℃焙烧3小时,865℃焙烧3小时得到成品催化剂,催化剂组成列于表1。
[0096]
按实施例1的方法评价和分析催化剂,测试结果列于表2。
[0097]
[实施例8]
[0098]
将相当于30.7份fe2o3的氧化铁红、相当于45.2份fe2o3的氧化铁黄、相当于5.05份k2o的碳酸钾、相当于8.84份ceo2的草酸铈、相当于2.58份wo3的钨酸铵、相当于1.38份sro的碳酸锶、1.15份geo2及5.69份石墨在捏合机中搅拌0.2小时,加入1.85份mnfe2o4和3.25份k2tio3,再搅拌1.8小时,加入占催化剂原料总重27.7%的脱离子水,拌和0.55小时,取出挤条,挤成直径3毫米、长6毫米的颗粒,放入烘箱,45℃烘2.5小时,85℃烘2.5小时,128℃烘3小时,然后置于马福炉中,于555℃焙烧2.5小时,675℃焙烧3小时,865℃焙烧3小时得到成品催化剂,催化剂组成列于表1。
[0099]
按实施例1的方法评价和分析催化剂,测试结果列于表2。不同的是,改变催化剂评价条件,即-50kpa、液体体积空速1.8小时-1
、反应温度575℃、水/乙苯重量比1.4。
[0100]
表1(待续)
[0101][0102]
表1(续)
[0103][0104]
表2
[0105][0106]
本发明所述实施例仅是对本发明技术方案的详细说明,但本发明不局限于上述实施例,即本发明不依赖上述实施例所述步骤才能实施。综上,本领域的技术人员对本发明进行的任何改进,包括对本发明所述原料及添加剂的替换、具体实施方式的选择等,均属于本发明的保护范围和公开范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1