一种聚酰胺复合反渗透膜及其制备方法与流程
1.本发明涉及纯水制造和废水处理的水处理技术领域,特别涉及一种具有高脱盐率、高渗透通量和高耐氧化性能的聚酰胺复合反渗透膜及其制备方法和应用。
背景技术:
2.目前市场上广泛销售的反渗透膜绝大部分是基于john.e.cadotte所发明的界面聚合法制备的交联芳香族聚酰胺复合膜(us4277344)。该复合膜的脱盐层是利用间苯二胺和均苯三甲酰氯在聚砜多孔支撑层的表面通过界面缩聚反应形成的交联芳香族聚酰胺。由于这种反渗透膜的脱盐率、渗透通量和耐酸碱性能较好,因而广泛应用于纯水制造、废水处理/回用和海水淡化等领域。然而,其在使用过程中面临着一些耐氧化性和脱盐层结合强度差等问题。
3.技术问题一:耐氧化性能差
4.根据市场的使用经验,反渗透膜在使用过程中会出现脱盐率下降的问题,这主要是由于水中的酸、碱或氧化性物质对芳香族聚酰胺脱盐层的破坏降解造成的。氧化性物质对聚酰胺脱盐层的破坏是快速且不可逆的,所以应极力避免氧化性物质与反渗透膜接触。次氯酸钠(活性成分以余氯浓度计)是反渗透膜进水中最广泛出现的氧化性物质,主要用来在预处理步骤中杀死微生物。为了控制反渗透膜进水的余氯降低至不破坏聚酰胺脱盐层的浓度(一般是在0.1ppm以下),还需要使用还原性的亚硫酸氢钠等消耗掉多余的次氯酸钠。当预处理出现波动时,过量次氯酸钠就进入到反渗透膜装置,氧化性的次氯酸钠会不可逆地降解聚酰胺脱盐层,使得膜脱盐率不可逆下降。因此,提升反渗透膜的耐氧化性能是十分必要且有价值的。
5.一般由间苯二胺和均苯三甲酰氯界面缩聚反应得到的交联芳香族聚酰胺脱盐层内存在的官能团有酰胺基、伯氨基和羧基,其中酰胺和伯氨基的活性氢很容易被氧化性氯取代而发生后续的聚合物降解。作为提升反渗透膜耐氧化性的方法,一般包括:1)通过添加或替换使用新的单体改变聚酰胺层结构;2)在聚酰胺层表面或内部接枝新的物质以提供“牺牲性”的氯攻击位点;3)化学浸渍后处理进入引入新的化学基团;4)在膜表面涂覆保护层。作为这些方法的实践,目前已经有了大量的工作。例如,中国发明专利cn100486682c和cn108430612a公开了使用过硫酸盐浸渍反渗透膜,并复配在油相中添加其它多元酰氯提升膜耐氯性能的方法,该方法处理后膜的脱盐率略微降低。中国发明专利cn109173742a、cn109794174a通过在膜表面涂布脂肪族醛、缩水甘油醚类物质、聚乙烯醇作为反渗透膜表面的交联保护层,进而提升了耐氯性能,然而保护层的引入不可避免地降低了膜的渗透通量。中国发明专利cn109603587a也通过将反渗透膜浸渍在硫代二乙酸或亚硫酸钠等还原性类物质,不仅提高了耐氯性能,还赋予了膜不易黄变发黑的性质。中国发明专利cn109647224a通过将界面缩聚形成的聚酰胺膜重新浸渍在高浓度的酰氯溶液中,使酰氯继续与聚酰胺内未反应的伯氨基反应,形成完全羧基封端的聚酰胺层,同样也提高膜的耐氧化性能。中国发明专利cn110975644a和cn111282458a通过化学接枝的方法,在反渗透膜表
面引入了多肽类物质、谷胱甘肽等含硫物质,作为活性氯的“牺牲”攻击点,明显提高了膜的耐氯性能。
6.目前市售的反渗透膜脱盐层的主体结构是交联芳香族聚酰胺,其包含的官能团有酰胺基、羧基和氨基。氧化性物质对聚酰胺层的破坏主要是针对酰胺基和氨基,且在破坏过程中伴随着自由基的产生和转移。因此,猝灭或减少聚酰胺结构中的自由基可以降低氧化性物质对脱盐层的破坏降解作用。作为举例,哌啶是一种含五个碳原子的环状仲胺化合物,具有猝灭自由基的作用,因而含有哌啶结构的聚合物具有良好的耐碱和耐氧化稳定性能,可以用作膜材料的制备。例如,刘品阳以溴代哌啶季铵盐、溴代二哌啶丙烷螺旋侧链盐等作为单体,制备了含有哌啶结构的碱性离子交换膜,该膜对氢氧化钠水溶液和芬顿试剂有良好的耐受性能(硕士学位论文:高性能杂环季铵盐接枝聚苯并咪唑碱性离子膜制备及性能研究[d].刘品阳.大连理工大学.2020);另外,金翠红合成了高分子量不含醚键的聚(间三联苯哌啶),再通过侧链功能化,制备了含哌啶鎓离子的碱性离子交换膜,同样对氢氧化钠水溶液和芬顿试剂有良好的耐受性能(博士学位论文:高效长侧链聚合物电解质膜制备及性能研究[d].金翠红.2019)。尽管如此,含有哌啶结构的聚合物本周的耐酸碱等耐化学性能较好,但是其很难在不影响反渗透膜通量脱盐率的前提下,将其引入到聚酰胺脱盐层上。
[0007]
以上提升反渗透膜耐氧化性能的方法,往往不可避免地降低脱盐率和渗透通量,很难做到在保持高脱盐率、高渗透通量的前提下,同时提升膜的耐氧化性能。
[0008]
技术问题二:脱盐层结合强度低
[0009]
除了上述提到的反渗透膜的耐氧化性性能,聚酰胺脱盐层与聚砜支撑层之间的结合强度的大小也直接影响到反渗透膜的运行过程的使用寿命。由于聚酰胺脱盐层与聚砜支撑层之间只是依靠“锚定”的物理作用结合在一起,当脱盐层未完全贴合在聚砜层表面时,在高膜面流速的酸碱冲洗时,很容易首先发生局部脱盐层受损或脱落,然后酸碱清洗液进入到脱盐层与聚砜层之间的结合部,进一步破坏聚酰胺层,从而造成膜脱盐率的不可逆下降。因此,通过配方和工艺手段,提升聚酰胺脱盐层与聚砜支撑层之间的结合强度具有实践价值。目前尚未有学术论文或专利专门研究如何提升聚酰胺层与聚砜层的结合强度。
技术实现要素:
[0010]
本发明提供一种同时满足高耐氧化性、高脱盐层结合强度、高脱盐率和高渗透通量的聚酰胺复合反渗透膜及其制备方法和应用。
[0011]
本发明的第一方面是提供一种聚酰胺复合反渗透膜,其包括无纺布、多孔支撑层和聚酰胺脱盐层,所述聚酰胺脱盐层中同时含有偶氮基、酚羟基和磺酸基,还至少含有2,2,6,6-四甲基哌啶结构和/或2,2,6,6-四甲基哌啶氮氧自由基结构。
[0012]
优选地,所述聚酰胺脱盐层是多元芳香族胺和多元芳香族酰氯通过界面缩聚反应形成的三维网状交联聚酰胺。
[0013]
优选地,所述2,2,6,6-四甲基哌啶结构或2,2,6,6-四甲基哌啶氮氧自由基结构是通过2,2,6,6-四甲基哌啶衍生物与聚酰胺脱盐层内未反应酰氯基团形成的化学键与三维网状交联聚酰胺相结合的。
[0014]
进一步优选地,所述2,2,6,6-四甲基哌啶衍生物选自以下物质中的至少一种:2,2,6,6-四甲基-4-哌啶醇、4-羟基-2,2,6,6-四甲基哌啶氮氧自由基、n-丁基-2,2,6,6-四甲
基-4-哌啶胺、4-氨基-2,2,6,6-四甲基哌啶、n,n-双(2,2,6,6-四甲基-4-哌啶胺)-1,6-己二胺。
[0015]
本发明的第二方面是提供一种聚酰胺复合反渗透膜的制备方法,包括如下步骤:
[0016]
(1)使用含多元芳香族胺和2,2,6,6-四甲基哌啶衍生物的水相溶液,以及含有多元芳香族酰氯的油相溶液,在所述包含无纺布和多孔支撑层的基膜表面进行界面缩聚反应,得到含2,2,6,6-四甲基哌啶结构和/或2,2,6,6-四甲基哌啶氮氧自由基结构的聚酰胺膜;
[0017]
(2)将步骤(1)得到的聚酰胺膜浸渍在含2,2,6,6-四甲基哌啶衍生物的热水溶液中;
[0018]
(3)使用热水对步骤(2)得到的聚酰胺膜进行清洗;
[0019]
(4)将步骤(3)得到的聚酰胺膜浸渍在亚硝酸水溶液中;
[0020]
(5)将步骤(4)得到的聚酰胺膜浸渍在亚硫酸钠水溶液中;
[0021]
(6)将步骤(5)得到的聚酰胺膜使用热水处理。
[0022]
优选地,以上所述步骤(1)水相溶液中2,2,6,6-四甲基哌啶衍生物的质量百分含量为0.1%~1%。
[0023]
优选地,以上所述步骤(2)热水溶液中2,2,6,6-四甲基哌啶衍生物的质量百分含量为0.01%~0.1%。
[0024]
优选地,以上所述步骤(3)得到的聚酰胺膜内多元芳香族胺的残留量小于5mg/m2。
[0025]
优选地,以上所述步骤(4)亚硫酸钠水溶液的质量百分含量为1.0%~5.0%。
[0026]
进一步优选地,以上所述步骤(2)、(3)、(6)中热水或热水溶液的温度范围是80~95℃。
[0027]
本发明的第三方面是提供一种聚酰胺复合反渗透膜的应用。作为耐氧化物聚酰胺复合反渗透膜的使用形式,不作任何限定,可以是螺旋卷式膜元件、板式膜组件和中空纤维膜组件的任一种。作为聚酰胺复合反渗透膜的应用领域,同样不作任何限制,例如超纯水制造、废水处理和海水淡化等。
[0028]
本发明所述的聚酰胺复合反渗透膜具有以下性能:在以25℃、浓度为2000mg/l氯化钠为原水,在1.55mpa操作压力下,反渗透膜的脱盐率在99.6%以上,渗透通量在50.0l/(m2·
h)以上,另外,膜片在25℃、ph为7、1000mg/l的次氯酸钠水溶液浸渍40小时后,脱盐率仍在98.0%以上。将膜片在3.8mpa下反压30min后,再在1.55mpa下测试,其脱盐率在98.5%以上,渗透通量在42.0l/(m2·
h)以上。
[0029]
以下对本发明内容进行具体说明。
[0030]
1.本发明第一方面:提供一种聚酰胺复合反渗透膜
[0031]
本发明的第一方面是聚酰胺复合反渗透膜,其包括无纺布、多孔支撑层构成的基膜,以及形成在基膜表面的交联芳香族聚酰胺脱盐层。
[0032]
1.1基膜
[0033]
基膜一般用作较薄的聚酰胺脱盐层的物理支撑层,赋予整个反渗透膜的机械强度,其本身不具备对小分子有机物和盐离子等的截留性能。基膜有无纺布基材和多孔支撑层两部分构成。下面分别进行说明。
[0034]
无纺布基材作为多孔支撑层的物理支撑材料,其作用是为可提供机械强度,并以
此为基础生成一层厚度、表面孔径均匀的多孔支撑层。
[0035]
作为无纺布基材的材质,可以优选聚乙烯、聚丙烯、聚对苯二甲酸乙二醇酯(简称pet),从亲水性、机械稳定性和热稳定等方面考虑,特别优选聚对苯二甲酸乙二醇酯。
[0036]
作为无纺布基材的厚度范围,优选在20~100μm范围内,更优选在80~90μm范围内。当无纺布厚度在60μm以下时,无纺布本身的强度不足以作为多孔支撑层的物理支撑,以此形成的基膜或反渗透膜有在横向或纵向破裂的风险,不能满足要求。当无纺布厚度在100μm以上时,虽然其作为物理支撑的强度足够,但是最后得到反渗透膜厚度太厚,一方面,造成由其制作的膜元件或组件的装填膜面积过低,影响到单只膜元件或组件的产水量,另一方面,在膜制备过程中无纺布基材内残留的未反应物质(主要是芳香族多元胺)的量增加,清洗困难,最后造成膜渗透通量降低,耐酸碱清洗性能也会下降。从物理支撑强度、膜元件或组件的装填膜面积和膜性能的角度考虑,更优选无纺布基材的厚度范围在80~90μm范围内。作为无纺布基材的厚度偏差范围优选在
±
5μm以下,更优选在
±
3μm以下。无纺布基材的厚度偏差过大时,在其表面使用刮刀或狭缝挤出(slot die)涂布用于多孔支撑层的铸膜液时,则比较困难,且会造成聚砜多孔支撑层厚度不均匀的问题。
[0037]
作为无纺布基材的透气度,优选在1.0~5.0cc/cm2/sec范围内,更优选在1.5~3.0cc/cm2/sec范围内,进一步优选在1.5~2.5cc/cm2/sec范围内。当透气度小于1.0cc/cm2/sec时,用于形成多孔支撑层的铸膜液在无纺布基材中的渗透深度很浅,造成多孔支撑层和无纺布基材之间的结合强度变弱,容易引起多孔支撑层从无纺布基材上的剥离。当透气度大于5.0时,用于形成多孔支撑层的铸膜液在无纺布基材中的渗透过大,甚至会渗透到无纺布基材的背面(非涂布面),造成针眼等涂布缺陷,也是非优选的。如果透气度在优选范围内,一方面多孔支撑层与无纺布基材的结合强度高,另一方面当涂布有用于形成多孔支撑层的铸膜液浸渍在凝固浴发生相转化时,凝固浴中非溶剂从背面(非涂布面)向铸膜液涂层的扩散速度合适,更容易形成非对称的多孔支撑层结构,对之后界面缩聚反应时多元胺的保持量和扩散速度也会造成有益效果。
[0038]
多孔支撑层作为聚酰胺脱盐层的物理支撑,夹在无纺布基材和聚酰胺脱盐层之间。
[0039]
作为多孔支撑层的材质,可以单独或混合地选自:聚砜、聚醚砜、磺化聚醚砜、线性芳香族聚酰胺、聚对苯二甲酸乙二醇酯、聚醚醚酮、聚苯砜、聚苯醚、聚乙烯、聚丙烯、聚丙烯腈、聚氯乙烯等。其中,更加优选聚砜、聚醚砜、聚丙烯腈、聚乙烯等。从表面张力、化学稳定性、刚性、热稳定性、溶解性等方面考虑,进一步优选聚砜。具体地说,使用聚砜材质制备多孔支撑层,则具有容易溶解在极性溶剂、形成支撑层耐压性好、易于控制表面孔径等优点,故特别优选。
[0040]
作为用于溶解聚砜材料的溶剂,可以单独或混合地选自:n,n-二甲基甲酰胺、n,n-二甲基乙酰胺、n-甲基吡咯烷酮、n-乙基基吡咯烷酮、1,3-二甲基-2-咪唑啉酮等。具体使用何种溶剂,只要能很好地溶解聚砜,且与凝固浴(如水)混溶即可。从成本、形成多孔支撑层孔的类型和易于操作型和安全性等方面考虑,优选n,n-二甲基甲酰胺或n,n-二甲基乙酰胺。
[0041]
作为多孔支撑层的厚度,优选10~60μm范围内,更优选在30~50μm范围内。
[0042]
需要指出的是,无纺布基材和多孔支撑层的厚度可通过数字式厚度测定仪,连续
测试15个样品,取平均值得到。多孔支撑层的厚度是将基膜厚度减去无纺布基材的厚度得到。
[0043]
作为多孔支撑层的表面孔径,优选在1~100nm范围内,更优选在5~50nm,进一步优选在7~20nm。表面孔径的测定采用场发射扫描电子显微镜(fe-sem),将基膜用导电胶粘贴在样品台上,对样品进行喷金处理后,拍摄10万倍下的电镜照片。根据fe-sem的配套软件标尺计算测量基膜表面孔径大小。
[0044]
1.2聚酰胺脱盐层
[0045]
聚酰胺脱盐层是反渗透膜中起到实际分离作用的功能层。
[0046]
优选地,所述聚酰胺脱盐层是多元芳香族胺和多元芳香族酰氯通过界面缩聚反应形成的三维网状交联聚酰胺。含有的基团种类有酰胺基、羧基和氨基。其中,酰胺基由酰氯和胺反应脱去一个分子的氯化氢得到,羧基由未反应的酰氯水解得到,氨基则来自于未反应的多元芳香族胺。此外,本发明的聚酰胺复合反渗透膜脱盐层中还同时含有偶氮基、酚羟基、磺酸基,以及2,2,6,6-四甲基哌啶结构和2,2,6,6-四甲基哌啶氮氧自由基结构中的一种或多种。
[0047]
本发明所述多元芳香族胺,是指分子中含有苯环结构和2个以上氨基的化合物,可单独或混合选自:间苯二胺、对苯二胺、邻苯二胺、3,5-二氨基苯甲酸、2,4二氨基苯磺酸、1,3,5-三氨基苯、n,n-二甲基间苯二胺、n,n-二乙基间苯二胺、3-氨基苄基胺、4-氨基苄基胺等。从易于获得和所得膜的脱盐率、渗透通量的角度,优选间苯二胺。
[0048]
本发明所述多元芳香族酰氯,是指分子中含有苯环结构和2个以上酰氯基团的化合物,可单独或混合选自:均苯三甲酰氯、间苯二甲酰氯、对苯二甲酰氯、联苯二甲酰二氯、偶氮苯二甲酰二氯。考虑到易于获得性和所得膜的脱盐率、渗透通量,优选均苯三甲酰氯。
[0049]
作为将偶氮基引入到聚酰胺脱盐层中的手段,不作任何限定,可举例将以上界面缩聚形成的聚酰胺与亚硝酸、四氧化二氮等接触,使聚酰胺未反应的芳香族胺发生重氮化反应,形成重氮盐基团,而后重氮盐基团继续与游离的芳香族胺继续偶联形成偶氮基。
[0050]
作为将酚羟基引入到聚酰胺层中的手段,不作任何限定地,可以直接将含有重氮盐的聚酰胺膜直接与含酚类化合物水溶液接触,使重氮盐与酚类化合物发生偶联反应引入酚羟基,也可以将含有重氮盐基团的聚酰胺膜浸渍在热水中,使重氮盐水解直接生成酚羟基。经本发明人深入研究发现,由于将含有重氮盐的聚酰胺膜浸渍的含有酚类化合物中时,发生的偶联反应使得聚酰胺层交联度过大,虽然进一步提高了膜的脱盐率,但是造成了渗透通量明显下降,故不优选;而采用直接加热使重氮盐水解产生酚羟基,对不存在交联度过大的问题,故优选。
[0051]
作为将磺酸基引入到聚酰胺层中的手段,优选将含有重氮盐基团的聚酰胺膜与含有亚硫酸钠溶液接触,使部分重氮盐基团被取代,从而在聚酰胺内部上引入磺酸基。
[0052]
作为将2,2,6,6-四甲基哌啶结构和/或2,2,6,6-四甲基哌啶氮氧化物结构引入到聚酰胺层的手段,不作任何限定,只要是通过化学键与聚酰胺层结合即可。例如,可以将含有2,2,6,6-四甲基哌啶结构或2,2,6,6-四甲基哌啶氮氧化物结构的“2,2,6,6-四甲基哌啶衍生物”添加到水相溶液中,使其一起参与到界面缩聚形成聚酰胺的过程中,也可以将含有2,2,6,6-四甲基哌啶衍生物的水溶液与新形成的聚酰胺脱盐层表面接触,利用表面未反应的酰氯基团与2,2,6,6-四甲基哌啶衍生物的氨基或羟基反应,将四甲基哌啶衍生物接枝到
聚酰胺层表面。这两种方法可以单独使用,也可以复配使用。从尽量提高聚酰胺层中2,2,6,6-四甲基哌啶结构和/或2,2,6,6-四甲基哌啶氮氧化物的数量角度看,优选两种方法复配使用。
[0053]
作为2,2,6,6-四甲基哌啶衍生物,不作任何限定,只要是分子中含有2,2,6,6-四甲基哌啶结构或2,2,6,6-四甲基哌啶氮氧化物结构,且包含氨基或羟基等易于与酰氯发生反应的基团即可。优选地,2,2,6,6-四甲基哌啶衍生物可以单独或混合选自:2,2,6,6-四甲基-4-哌啶醇、4-羟基-2,2,6,6-四甲基哌啶氮氧自由基、n-丁基-2,2,6,6-四甲基-4-哌啶胺、4-氨基-2,2,6,6-四甲基哌啶、n,n-双(2,2,6,6-四甲基-4-哌啶胺)-1,6-己二胺。
[0054]
作为2,2,6,6-四甲基哌啶结构和/或2,2,6,6-四甲基哌啶氮氧化物结构对聚酰胺脱盐层性能有益效果的机理,尽管不希望受到理论的束缚,经本发明人深入研究发现,2,2,6,6-四甲基哌啶中4个取代甲基的强烈空间位阻效应改变了哌啶环上仲胺基的化学性质,赋予了其捕获或猝灭自由基的能力,而且2,2,6,6-四甲基哌啶被氧化后,形成可稳定存在的2,2,6,6-四甲基哌啶氮氧自由基结构,其具有猝灭单线态氧的性质,所以2,2,6,6-四甲基哌啶或氮氧自由基的引入有效提升了反渗透膜的耐氧化性能。此外,相比间苯二胺,2,2,6,6-四甲基哌啶结构还更容易夺取反渗透产生的氯化氢,从而使得界面缩聚反应最初产生的聚酰胺分子量更大,反应也更为充分,形成更多的聚酰胺,充分充满聚砜支撑层表面的表面孔,从而为形成的聚酰胺脱盐层提供更为牢固的“锚定”位点,进而提升了聚酰胺脱盐层与聚砜支撑层的结合强度。
[0055]
为了方便后续制作膜元件,作为可选,可选择制造干燥的反渗透膜。为了防止膜片在干燥过程中多孔支撑层出现坍塌而使得膜渗透通量降低,需使用保湿剂对膜片进行保湿。作为用于本发明反渗透膜的保湿剂,不作任何限定,只要是可吸湿性的物质、不破坏反渗透膜材料即可,可举例如甘油、柠檬酸、乙酸钠、葡萄糖等。
[0056]
另外,为了减少膜元件制作过程中的机械划伤,作为可选,可选择使用亲水性聚合物作为物理保护层,涂布在聚酰胺脱盐层的表面。作为亲水性聚合物,不作特别地限定,只要在干燥时有一定机械强度以起到保护作用,且很容易在膜元件使用过程中被水流冲走即可,举例如聚乙烯醇、聚氧乙烯、羧甲基纤维素、聚乙烯吡咯烷酮等。
[0057]
2.提供一种聚酰胺复合反渗透膜的制造方法
[0058]
接下来,对上述聚酰胺复合反渗透膜的制造方法进行详细描述。制造方法包括基膜的制造步骤和聚酰胺脱盐层的制造步骤。
[0059]
2.1基膜的制造步骤
[0060]
聚砜基膜的制造步骤包括:(1)在无纺布基材上涂布聚砜铸膜液,(2)浸渍在凝固浴中相转化固化成膜,(3)清洗。下面分别进行说明。
[0061]
步骤(1):在无纺布基材上涂布聚砜铸膜液
[0062]
关于聚砜铸膜液的固含量,以质量百分比计,优选在14~20%,更优选在15~18%,进一步优选在16~17%。关于聚砜铸膜液的温度,优选为10~50℃,更优选为15~30℃,进一步优选为20~25℃。如果聚砜铸膜液的固含量和温度在该范围内,则聚砜多孔支撑层会牢固地与无纺布基材相结合,且表面孔径在优选范围内。需要指出的是,聚砜多孔支撑层的孔径大小可以通过铸膜液的固含量和温度进行调节。
[0063]
关于聚砜铸膜液在无纺布基材上的涂布方式,不作任何限定,只要将铸膜液均匀
涂布到无纺布基材上即可。可以举例采用v字型刮刀或狭缝挤出(slot die)涂布的方式,从易于操作性和维护难度角度考虑,优选狭缝挤出(slot die)涂布的方式。
[0064]
步骤(2):浸渍在凝固浴中相转化固化成膜
[0065]
在无纺布基材上涂布完铸膜液之后、到接触到凝固浴之前在空气中的时间优选在0.5~10秒的范围内。当在空气中停留时间过长时,空气中的水蒸气会进入铸膜液与空气接触面而发生过多的预凝胶作用,导致聚砜多孔支撑层表面孔径过大且分布不均匀。带有铸膜液涂层的无纺布进入凝固浴之后、涂布面第一次接触导辊之前的时间优选在5秒以上,更优选在10秒以上,进一步优选在15秒以上,当该停留时间过短时,聚砜铸膜液还未来得及完全固化便接触导辊,会有多孔支撑层受到导辊挤压被破坏的风险。
[0066]
作为凝固浴,不做特别限定,只要能与二甲基甲酰胺互溶,且不能溶解聚砜材料即可,可以优选水、甲醇、乙醇、异丙醇、丙酮等。从安全性、成本和易于操作性考虑,优选为水。为了控制铸膜液在凝固浴中的相转化速度,作为优选,可以在凝固浴水中添加与铸膜液相同的溶剂二甲基甲酰胺。关于凝固浴中二甲基甲酰胺的浓度,以质量百分比计,优选在40%以下,更优选在30%以下,进一步优选在20%以下。当凝固浴中二甲基甲酰胺浓度过高时,铸膜液的相转化速度变慢,使得生产变得难以进行。关于凝固浴的温度,优选在0~40℃范围内,更优选在5~20℃范围内,进一步优选在10~15℃范围内。当凝固浴温度低于5℃时,铸膜液相转化速度变慢,不利于提高生产线效率;当凝固浴温度高于40℃时,相转化速度过于剧烈,使得形成的聚砜多孔支撑层表面孔径分布不均匀,同时凝固浴中的二甲基甲酰胺溶剂挥发也增加,不利于生产环境的改善。
[0067]
步骤(3):清洗
[0068]
在凝固浴中相转化固化形成含聚砜多孔支撑层的基膜后,还需要使用纯水清洗掉基膜内部残留的二甲基甲酰胺溶剂。关于清洗用纯水的温度,优选在40~100℃范围内,更优选在60~95℃范围内,进一步优选在70~85℃范围内。当清洗用纯水的温度在优选范围内,基膜内二甲基甲酰胺向外扩散的速度更快,清洗的更为充分。关于清洗时间,优选在1~10分钟,进一步优选为2~3分钟。为了达到充分的清洗效果,作为优选,可以将基膜依次通过若干个清洗水槽。作为清洗完毕后基膜内二甲基甲酰胺的残留量,优选在200mg/m2以下,进一步优选在50mg/m2以下。需要指出的是,基膜中二甲基甲酰胺的残留量的测试方法是:将100平方厘米的基膜膜片,剪碎后浸渍在50ml、80℃含20%的乙醇水溶液,在密封条件下静置2小时,待冷却至室温后,利用色谱测试浸取液中二甲基甲酰胺的浓度,换算出每平方米基膜中二甲基甲酰胺的残留量。
[0069]
2.2聚酰胺脱盐层的制造步骤
[0070]
在基膜表面形成聚酰胺脱盐层的制造步骤包括:(1)在基膜上形成聚酰胺;(2)2,2,6,6-四甲基哌啶衍生物热水溶液浸渍;(3)热水清洗;(4)亚硝酸水溶液浸渍;(5)亚硫酸钠水溶液浸渍;(6)热水清洗。
[0071]
下面进行详细说明。
[0072]
步骤(1):在基膜上形成聚酰胺。
[0073]
(1-a)首先,将基膜表面与多元芳香族胺和2,2,6,6-四甲基哌啶衍生物水相溶液接触,使其浸润基膜表面孔。
[0074]
此处多元芳香族胺浓度,以质量百分比计,优选在0.5%~10%,更优选在1%~
7%。关于水相溶液的温度,优选在0~100℃,更优选在5~60℃,进一步优选在20~45℃。
[0075]
关于2,2,6,6-四甲基哌啶衍生物在水相中的浓度,以质量百分比计,优选为0.1%~1.0%。当其浓度低于0.1%,引入到聚酰胺内的2,2,6,6-四甲基哌啶衍生物的量过少,无法起到足够的耐氧化作用;当其浓度大于1.0%时,2,2,6,6-四甲基哌啶衍生物对缩聚反应的妨碍过大,其氨基或羟基导致聚酰胺链段过早封端,使得聚酰胺脱盐层的交联度降低。
[0076]
为了促进水相溶液在基膜表面的铺展,作为可选,可在水相溶液中添加十二烷基硫酸钠、十二烷基苯磺酸钠、聚乙烯吡咯烷酮等表面活性剂。另外,只要不妨碍界面缩聚反应,还可以在水相溶液添加酰化催化剂、极性溶剂、缚酸剂和抗氧化剂等。关于酰化催化剂,可列举如己内酰胺、吡啶、4-二甲氨基吡啶等;关于极性溶剂,可列举如n-甲基吡咯烷酮、二甲基甲酰胺、乙醇、甲醇、异丙醇、二甲基亚砜、六甲基磷酰胺等;关于缚酸剂,可列举氢氧化钠、碳酸钠、碳酸氢钠、磷酸三钠、n,n-二甲基哌嗪等;关于抗氧化剂,可列举亚硫酸钠、焦亚硫酸钠、抗坏血酸等。
[0077]
关于含多元芳香族胺和2,2,6,6-四甲基哌啶衍生物的水相溶液与基膜表面接触的方法,只要能使水相溶液与基膜表面均匀接触即可,可通过涂布的方式将水相溶液均匀涂布在基膜表面,也可以将基膜直接浸渍在水相溶液中。关于水相溶液与基膜的接触时间,优选在5秒~10分钟,更优选在10秒~2分钟。接着,需要将基膜表面多余的水相溶液去除。作为去除方法,不作任何限定,只要能使水相溶液在基膜表面均匀浸润,且基膜表面没有肉眼可见的水珠即可。可以采用将基膜竖直使水相溶液自然流下而后自然干燥的方法,也可直接采用挤压辊或风刀先将大部分多余水相溶液除去,而后经自然晾干或烘箱干燥的方式继续除去剩余的水相溶液的方法。
[0078]
(1-b)然后,将多元芳香族酰氯的油相溶液均匀涂布在基膜表面,静态接触反应形成聚酰胺脱盐层。
[0079]
关于静态接触反应时间优选在0.5秒~5分钟,更优选在5秒~2分钟,进一步优选在10秒~1分钟。当静态接触反应时间低于0.5秒时,界面缩聚反应不充分,无法形成完整的聚酰胺脱盐层;当静态接触反应时间高于5分钟,则形成的聚酰胺脱盐层过厚,膜渗透通量过低,且造成生产效率的降低。
[0080]
关于油相溶液中多元芳香族酰氯的浓度,以质量百分比计,优选在0.05%~1%范围内,更优选在0.08%~0.5%范围内,进一步优选在0.1%~0.3%范围内。
[0081]
溶解多元芳香族酰氯的油相溶剂不作特别限定,只要与水不互溶、可形成油水界面、且不会溶解多孔支撑层材料和无纺布基材即可,如苯类、卤代烃类和烷烃类溶剂均可。作为苯类的溶剂,可以举例如苯、甲苯等。作为卤代烃类溶剂,可以举例如三氯三氟乙烷。作为烷烃类溶剂,可以举例如正己烷、正庚烷、正癸烷、正十一烷、正十二烷、正十三烷、正十四烷、正十七烷等直链烷烃,也可以举例如环辛烷、乙基环己烷等环烷烃,也可以举例如埃克森
·
美孚公司生产的isopar e、isopar g、isoapar l、isopar m等异构烷烃。
[0082]
(1-c)接下来,需要将膜表面多余的油相溶液去除。
[0083]
关于油相溶液的去除方法,不作任何限定,只要不破坏聚酰胺脱盐层即可。例如,可以采用直接使用烘箱使油相溶剂挥发的方式,还可以采用将膜竖直使油相溶液自然流下而后自然晾干的方式,也可以采用导辊、风刀、水刀等物理方法直接除去溶剂的方式。
[0084]
由此,可以得到内部含2,2,6,6-四甲基哌啶结构和/或2,2,6,6-四甲基哌啶氮氧
化物结构的聚酰胺膜。
[0085]
步骤(2):2,2,6,6-四甲基哌啶衍生物热水溶液浸渍。
[0086]
此步骤的目的是使2,2,6,6-四甲基哌啶衍生物的氨基或羟基与步骤(1)得到聚酰胺膜表面未反应的酰氯基团反应,使其接枝到聚酰胺膜表面。关于2,2,6,6-四甲基哌啶衍生物的热水溶液的浓度,以质量百分比计,优选在0.01%~0.1%范围内。关于热水溶液的温度,优选在70~100℃范围内,更优选在80~95℃范围内,如果热水溶液温度低于70℃,则会有2,2,6,6-四甲基哌啶衍生物向聚酰胺膜表面扩散较慢而导致反应不充分的问题;如果热水溶液温度高于100℃,则会有生产难以操作的问题。关于膜在热水溶液中浸渍的时间,优选在1秒~5分钟,更优选在10秒~2分钟,进一步优选在30秒~1分钟。
[0087]
步骤(3):热水清洗。
[0088]
此步骤的主要目的是将膜内残留的多元芳香族胺和在步骤(1)引入的2,2,6,6-四甲基哌啶衍生物清洗出来。关于清洗用纯水的温度,优选在60~100℃范围内,更优选在80~95℃范围,若温度过低则存在清洗不充分的问题。关于清洗时间,优选在30秒~10分钟,更优选在1分钟~3分钟,如果清洗时间低于30秒,则存在清洗不充分的问题,如果清洗时间大于10分钟,则存在膜渗透通量下降过多的问题。为了进一步提升清洗效果,作为可选,可以将膜依次通过若干个热水槽进行清洗。关于清洗完毕后膜内多元芳香族胺的残留量,优选在5mg/m2以下。如果膜内多元芳香族胺的残留量大于5mg/m2,则会在后续与亚硝酸形成重氮盐的进行偶联反应,造成聚酰胺脱盐层的交联度过大,造成膜的渗透通量降低较多。
[0089]
步骤(4):亚硝酸水溶液浸渍。
[0090]
此步骤的主要目的是使聚酰胺脱盐层内的芳香族胺与亚硝酸发生重氮化反应生成重氮盐基团,而后,该重氮盐与膜内残留的极少量(5mg/m2以下)多元芳香族胺以及聚酰胺链段相邻的氨基发生偶联反应,在聚酰胺脱盐层内引入偶氮基。需要指出的是,一方面由于膜内残留的游离多元芳香族胺的量极低(5mg/m2以下),另一方面在室温或更低温度下,聚酰胺链段内未反应氨基活动性较差,所以此步骤引入的偶氮基数量较少,大部分的重氮盐基团得到保留,从而使得聚酰胺脱盐层不会被偶氮基过度交联而造成渗透通量降低。
[0091]
聚酰胺膜内多元芳香族胺的残留量的测试方法为:将10张面积为28.32平方厘米的圆形膜片剪碎,密封浸泡在80℃、含20%乙醇的水溶液中,取浸取液,使用紫外分光光光度计测试浸取液中多元芳香族胺的浓度,然后换算得到每平方米中多元芳香族胺的残留量。
[0092]
关于亚硝酸水溶液的浓度,以质量百分比计,优选在0.01%~1.0%范围内,进一步优选在0.1%~0.5%范围内。如果浓度低于0.01%,则聚酰胺脱盐层内的芳香族胺的重氮盐反应不充分;如果浓度高于1%,则亚硝酸会分解产生氮氧化物气体,生产变得难以操作。关于亚硝酸水溶液的温度,优选在0~40℃范围内,更优选在5~30℃,进一步优选在10~20℃。当亚硝酸水溶液温度低于0℃时,亚硝酸水溶液容易有结冰风险而破坏聚酰胺膜的风险,当亚硝酸水溶液温度高于40℃时,亚硝酸分解产生氮氧化物气体,使得生产变得困难。尽管可以直接使用亚硝酸,但更优选使用亚硝酸盐与酸反应生成的新配制的溶液。亚硝酸盐可举例如亚硝酸钠,与之反应的酸不作任何限定,可以是盐酸、硫酸、硝酸、硫酸等无机酸,也可是柠檬酸、乙酸、乙基磺酸、甲基磺酸、三氟甲磺酸等有机酸,从经济性和易于操作性角度看,优选使用盐酸或硫酸与亚硝酸钠反应直接形成亚硝酸水溶液。关于亚硝酸水溶
液的ph值,优选在7以下,更优选在5以下,进一步优选在3以下,为了防止亚硝酸水溶液由于酸性过强导致亚硝酸分解产生气体速度过快,水溶液ph应在1以上。
[0093]
聚酰胺膜浸渍在亚硝酸水溶液中的时间不作特别限定,只要能使聚酰胺脱盐层内未反应芳香族胺与亚硝酸充分反应即可。该重氮化反应所需的接触时间与亚硝酸水溶液的中hno2分子的浓度有关,浓度越高所需的接触时间就越短。根据以上优选的亚硝酸水溶液的浓度、温度和ph范围,该接触时间优选在5秒~10分钟,更优选在10秒~3分钟,进一步优选在30秒~1分钟。另外,只要不妨碍亚硝酸与芳香族胺的重氮化反应,还可以在亚硝酸水溶液中添加表面活性剂以促进亚硝酸向聚酰胺膜内的扩散,所需表面活性剂可列举十二烷基硫酸钠、十二烷基苯磺酸钠、聚乙烯吡咯烷酮等。
[0094]
由此,在聚酰胺脱盐层中引入了重氮盐基团和少量的偶氮基。
[0095]
步骤(5):亚硫酸钠水溶液浸渍。
[0096]
此步骤的主要目的是使用亚硫酸钠与聚酰胺脱盐层内的重氮盐基团发生取代反应,在聚酰胺层内引入磺酸基。关于亚硫酸钠水溶液的浓度,以质量百分比计,优选在0.1%~20%范围内,更优选在1%~10%范围内,进一步优选在1%~5%范围内。如果亚硫酸钠浓度在此优选范围内,一方面可以使重氮盐基团与亚硫酸钠中亚硫酸根离子充分反应,另一方面还可以在后续热水处理上减轻操作难度。关于亚硫酸钠水溶液的温度,优选在10~60℃范围内,更优选在20~45℃范围内,进一步优选在25~35℃范围内。关于膜与亚硫酸钠水溶液接触时间,优选在5秒~5分钟,更优选在10秒~2分钟,进一步优选在30秒~1分钟。
[0097]
由此,在聚酰胺脱盐层内引入了磺酸基。
[0098]
步骤(6):热水清洗。
[0099]
此步骤的主要目的是使聚酰胺内部残留重氮盐基团加热充分水解被取代为酚羟基,此外,还可对膜内残留的亚硫酸钠进行清洗。关于热水的温度,优选在60~100℃范围内,进一步优选在80~95℃范围内。当热水温度低于60℃时,重氮盐的水解反应则变慢,酚羟基的生成也变得困难。关于膜在热水中的处理时间,优选在5秒~10分钟,更优选在30秒~5分钟,进一步优选在1分钟~2分钟。处理时间过短时,重氮盐的水解不充分,而处理时间过长,容易造成聚酰胺膜渗透通量的下降。
[0100]
作为可选,在制造干燥反渗透膜片时,可以使用保湿剂对反渗透膜进行处理而后再在烘箱中干燥的方法。作为使用保湿剂处理反渗透膜的手段,可以直接将反渗透膜浸渍在保湿剂水溶液中,也可以仅将反渗透膜背面(无纺布基材面)与保湿剂水溶液接触的方法。保湿剂水溶液的浓度,以质量百分比计,优选在3.5~20%范围内,如果保湿剂浓度在此优选范围内,则不仅可以使反渗透膜多孔支撑层内的孔充分填充,而且在成本上也是可以接受的。
[0101]
作为可选,为了减少膜元件制造过程中聚酰胺层所收到的物理划伤,可以在聚酰胺脱盐层表面涂布以上涂布亲水性聚合物物理保护层。作为涂布方式,可以选择任何公知的技术,例如辊涂、狭缝挤出(slot die)涂布等方式。作为亲水性聚合物的浓度,以质量百分比计,优选在0.05%~5%范围内。
[0102]
3.本发明的第三方面:提供聚酰胺复合反渗透膜的应用
[0103]
为了方便使用,可以将所述耐氧化聚酰胺反渗透膜制造在膜元件或膜组件,其形式不做任何限定,可以是螺旋卷式膜元件、板式膜组件和中空纤维膜组件的任一种。可以使
用本发明的聚酰胺复合反渗透膜或膜元件/组件处理的原水包括但不限于纯水制造、废水处理/回用和海水淡化,原水总溶解性固体含量tds范围在500~50000mg/l。
[0104]
本发明的有益效果如下:本发明提供的耐氧化反渗透膜及其制备方法在提高反渗透耐氧化性、高脱盐层结合强度的同时,仍保持了高的渗透通量和脱盐率。
[0105]
在以25℃、浓度为2000mg/l氯化钠为原水,在1.55mpa操作压力下,反渗透膜的脱盐率在99.6%以上,渗透通量在50.0l/(m2·
h)以上,另外,膜片在25℃、ph为7、1000mg/l的次氯酸钠水溶液浸渍40小时后,脱盐率仍在98.0%以上;将膜片在3.8mpa下反压30min后,再在1.55mpa下测试,其脱盐率在98.5%以上,渗透通量在42.0l/(m2·
h)以上。另外,本发明的工艺容易实现产业化。
具体实施方式
[0106]
为了更好地理解本发明的具体内容,下面结合实施例进一步阐述本发明的内容,但本发明的内容不仅仅局限于以下实施例。
[0107]
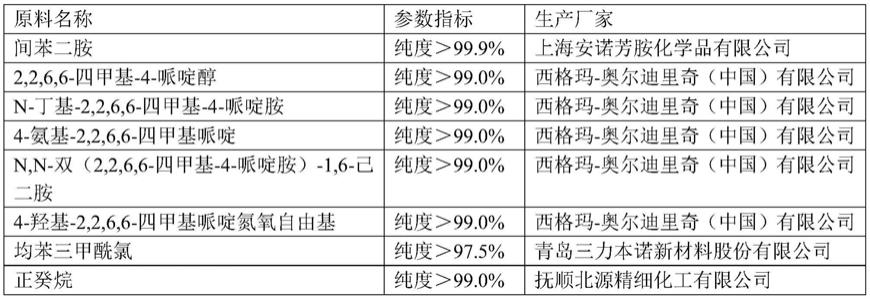
以下实施例或对比例中所使用的原料若未特别说明,均为市售常规原料,主要原料信息见下表1:
[0108]
表1主要原料来源
[0109][0110][0111]
以下本发明实施例或对比例中可能用到的方法进行说明:
[0112]
1.脱盐率和渗透通量的评价
[0113]
脱盐率和渗透通量是评价反渗透膜分离性能的两个重要参数。本发明根据gb/t 32373-2005《反渗透膜测试方法》中的内容对反渗透膜进行分离性能评价。
[0114]
脱盐率(r)定义为:在一定操作条件下,反渗透膜进料液盐浓度(cf)与渗透液盐浓度(c
p
)之差,再除以进料液盐浓度(cf),如下公式所述。
[0115][0116]
渗透通量的定义为:在一定操作条件下,单位时间内透过单位膜面积的水的体积,
4-哌啶胺外,与实施例1同样地制造反渗透膜。对得到聚酰胺复合反渗透膜的分离性能、2号水槽后与3号水槽前的膜内间苯二胺的残留量、耐氧化性能、脱盐层结合强度进行评价,结果见表2。
[0129]
实施例3
[0130]
除将实施例1中的4-氨基-2,2,6,6-四甲基哌啶替换为2,2,6,6-四甲基-4-哌啶醇外,与实施例1同样地制造反渗透膜。对得到聚酰胺复合反渗透膜的分离性能、2号水槽后与3号水槽前的膜内间苯二胺的残留量、耐氧化性能、脱盐层结合强度进行评价,结果见表2。
[0131]
实施例4
[0132]
除将实施例1中的4-氨基-2,2,6,6-四甲基哌啶替换为n,n-双(2,2,6,6-四甲基-4-哌啶胺)-1,6-己二胺外,与实施例1同样地制造反渗透膜。对得到聚酰胺复合反渗透膜的分离性能、2号水槽后与3号水槽前的膜内间苯二胺的残留量、耐氧化性能、脱盐层结合强度进行评价,结果见表2。
[0133]
实施例5
[0134]
除将实施例1中的4-氨基-2,2,6,6-四甲基哌啶替换为4-羟基-2,2,6,6-四甲基哌啶氮氧自由基外,与实施例1同样地制造反渗透膜。对得到聚酰胺复合反渗透膜的分离性能、2号水槽后与3号水槽前的膜内间苯二胺的残留量、耐氧化性能、脱盐层结合强度进行评价,结果见表2。
[0135]
对比例1
[0136]
除了将水相溶液和1号水槽不添加4-氨基-2,2,6,6-四甲基哌啶外,与实施例1同样地制造反渗透膜。对得到聚酰胺复合反渗透膜的分离性能、2号水槽后与3号水槽前的膜内间苯二胺的残留量、耐氧化性能、脱盐层结合强度进行评价,结果见表2。
[0137]
表2实施例1-5及对比例1膜性能
[0138][0139]
从实施例1~5和对比例1可以看出,通过在反渗透膜制造过程中引入四甲基哌啶衍生物,反渗透膜的耐氯性能和脱盐层结合强度均有明显提高。另外,2号水槽后与3号水槽前的膜内间苯二胺残留量均在5mg/m2以下,实施例1~5中的反渗透膜片初始脱盐率均在99.6%以上,渗透通量均在50.0l/(m2·
h)以上。对比例1中未使用四甲基哌啶衍生物的反渗透膜经次氯酸钠处理后,脱盐率低于98.0%;经反压后其脱盐率低于98.5%,通量低于42.0l/(m2·
h),表现出较差的耐次氯酸钠氧化性能和脱盐层结合强度。
[0140]
实施例6
[0141]
除了将水相溶液4-氨基-2,2,6,6-四甲基哌啶的质量百分比由0.2%改为0.1%外,与实施例1同样地制造反渗透膜。对得到聚酰胺复合反渗透膜的分离性能、2号水槽后与
3号水槽前的膜内间苯二胺的残留量、耐氧化性能、脱盐层结合强度进行评价,结果见表3。
[0142]
实施例7
[0143]
除了将水相溶液4-氨基-2,2,6,6-四甲基哌啶的质量百分比由0.2%改为0.5%外,与实施例1同样地制造反渗透膜。对得到聚酰胺复合反渗透膜的分离性能、2号水槽后与3号水槽前的膜内间苯二胺的残留量、耐氧化性能、脱盐层结合强度进行评价,结果见表3。
[0144]
实施例8
[0145]
除了将水相溶液质量百分比为0.2%的4-氨基-2,2,6,6-四甲基哌啶改为由质量百分比为0.2%的2,2,6,6-四甲基-4-哌啶醇与质量百分比为0.3%的4-羟基-2,2,6,6-四甲基哌啶氮氧自由基外,与实施例1同样地制造反渗透膜。对得到聚酰胺复合反渗透膜的分离性能、2号水槽后与3号水槽前的膜内间苯二胺的残留量、耐氧化性能、脱盐层结合强度进行评价,结果见表3。
[0146]
对比例2
[0147]
除了将水相溶液4-氨基-2,2,6,6-四甲基哌啶的质量百分比由0.2%改为1.5%外,与实施例1同样地制造反渗透膜。对得到聚酰胺复合反渗透膜的分离性能、2号水槽后与3号水槽前的膜内间苯二胺的残留量、耐氧化性能、脱盐层结合强度进行评价,结果见表3。
[0148]
表3实施例6-8及对比例2膜性能
[0149][0150]
从实施例6~8和对比例2可以看出,在水相溶液中添加不同浓度的一种或两种四甲基哌啶衍生物,反渗透膜的耐氯性能和脱盐层结合强度均有明显提高。另外,2号水槽后与3号水槽前的膜内间苯二胺残留量均在5mg/m2以下,实施例6~8的反渗透膜片初始脱盐率均在99.6%以上,初始渗透通量均在50.0l/(m2·
h)以上。然而,对比例2中由于在水相溶液中四甲基哌啶衍生物添加量过多,导致反渗透膜的初始脱盐率低于99.6%,3.8mpa反压后脱盐率低于98.5%,表现出较低的初始脱盐率和脱盐层结合强度。
[0151]
实施例9
[0152]
除了将1号水槽中4-氨基-2,2,6,6-四甲基哌啶的质量百分比由0.05%改为0.01%外,与实施例1同样地制造反渗透膜。对得到聚酰胺复合反渗透膜的分离性能、2号水槽后与3号水槽前的膜内间苯二胺的残留量、耐氧化性能、脱盐层结合强度进行评价,结果见表4。
[0153]
实施例10
[0154]
除了将1号水槽中4-氨基-2,2,6,6-四甲基哌啶的质量百分比由0.05%改为0.1%外,与实施例1同样地制造反渗透膜。对得到聚酰胺复合反渗透膜的分离性能、2号水槽后与3号水槽前的膜内间苯二胺的残留量、耐氧化性能、脱盐层结合强度进行评价,结果见表4。
[0155]
对比例3
[0156]
除了将1号水槽中4-氨基-2,2,6,6-四甲基哌啶的质量百分比由0.05%改为0.15%外,与实施例1同样地制造反渗透膜。对得到聚酰胺复合反渗透膜的分离性能、2号水槽后与3号水槽前的膜内间苯二胺的残留量、耐氧化性能、脱盐层结合强度进行评价,结果见表4。
[0157]
表4实施例9-10及对比例3膜性能
[0158][0159]
从实施例9~10和对比例3可以看出,改变1号水槽中四甲基哌啶衍生物的浓度,2号水槽后与3号水槽前的膜内间苯二胺残留量均在5mg/m2以下,实施例9~10的反渗透膜片初始脱盐率均在99.6%以上,初始渗透通量均在50.0l/(m2·
h)以上。然而,对比例3中由于第1个水槽四甲基哌啶衍生物添加量过多,导致反渗透膜的通量低于50.0l/(m2·
h),表现出较低的初始通量。
[0160]
实施例11
[0161]
除了将第1、2号水槽温度由90℃改为80℃,与实施例1同样地制造反渗透膜。对得到聚酰胺复合反渗透膜的分离性能、2号水槽后与3号水槽前的膜内间苯二胺的残留量、耐氧化性能、脱盐层结合强度进行评价,结果见表5。
[0162]
实施例12
[0163]
除了将第1、2号水槽温度由90℃改为85℃,与实施例1同样地制造反渗透膜。对得到聚酰胺复合反渗透膜的分离性能、2号水槽后与3号水槽前的膜内间苯二胺的残留量、耐氧化性能、脱盐层结合强度进行评价,结果见表5。
[0164]
实施例13
[0165]
除了将第1、2号水槽温度由90℃改为95℃,与实施例1同样地制造反渗透膜。对得到聚酰胺复合反渗透膜的分离性能、2号水槽后与3号水槽前的膜内间苯二胺的残留量、耐氧化性能、脱盐层结合强度进行评价,结果见表5。
[0166]
实施例14
[0167]
除了将5号水槽温度由90℃改为80℃,与实施例1同样地制造反渗透膜。对得到聚酰胺复合反渗透膜的分离性能、2号水槽后与3号水槽前的膜内间苯二胺的残留量、耐氧化性能、脱盐层结合强度进行评价,结果见表5。
[0168]
实施例15
[0169]
除了将5号水槽温度由90℃改为95℃,与实施例1同样地制造反渗透膜。对得到聚酰胺复合反渗透膜的分离性能、2号水槽后与3号水槽前的膜内间苯二胺的残留量、耐氧化性能、脱盐层结合强度进行评价,结果见表5。
[0170]
对比例4
[0171]
除了将第1、2号水槽温度由90℃改为60℃,与实施例1同样地制造反渗透膜。对得到聚酰胺复合反渗透膜的分离性能、2号水槽后与3号水槽前的膜内间苯二胺的残留量、耐
氧化性能、脱盐层结合强度进行评价,结果见表5。
[0172]
对比例5
[0173]
除了将5号水槽温度由90℃改为25℃,与实施例1同样地制造反渗透膜。对得到聚酰胺复合反渗透膜的分离性能、2号水槽后与3号水槽前的膜内间苯二胺的残留量、耐氧化性能、脱盐层结合强度进行评价,结果见表5。
[0174]
表5实施例11-15及对比例4-5膜性能
[0175][0176]
从实施例11~15和对比例5可以看出,改变第1、2、5号水槽温度在80~95℃范围内,2号水槽后与3号水槽前的膜内间苯二胺残留量均在5mg/m2以下,实施例11~15的反渗透膜片初始脱盐率均在99.6%以上,初始渗透通量均在50.0l/(m2·
h)以上。然而,对比例4中由于第1、2号水槽温度较低,间苯二胺未清洗干净,膜内间苯二胺残留量高于5mg/m2,导致反渗透膜的初始渗透通量低于50.0l/(m2·
h)。对比例5中由于第5个水槽温度不够,重氮盐水解产生酚羟基的反应受到了抑制,故反渗透膜的初始渗透通量也低于50l/(m2·
h),1000ppm次氯酸钠浸泡40h后,其脱盐率低于98.5%,经3.8mpa反压后其脱盐率低于98.5%,通量低于42.0l/(m2·
h),表现出较差的初始分离性能、耐次氯酸钠氧化性能和脱盐率结合强度。
[0177]
实施例16
[0178]
除了将4号水槽温度亚硫酸钠浓度由3%改为5%,与实施例1同样地制造反渗透膜。对得到聚酰胺复合反渗透膜的分离性能、2号水槽后与3号水槽前的膜内间苯二胺的残留量、耐氧化性能、脱盐层结合强度进行评价,结果见表6。
[0179]
实施例17
[0180]
除了将4号水槽温度亚硫酸钠浓度由3%改为1%,与实施例1同样地制造反渗透膜。对得到聚酰胺复合反渗透膜的分离性能、2号水槽后与3号水槽前的膜内间苯二胺的残留量、耐氧化性能、脱盐层结合强度进行评价,结果见表6。
[0181]
对比例6
[0182]
除了将4号水槽温度亚硫酸钠浓度由3%改为0.1%,与实施例1同样地制造反渗透膜。对得到聚酰胺复合反渗透膜的分离性能、2号水槽后与3号水槽前的膜内间苯二胺的残留量、耐氧化性能、脱盐层结合强度进行评价,结果见表6。
[0183]
表6实施例15-16及对比例6膜性能
[0184][0185]
从实施例16~17和对比例6可以看出,改变4号水槽亚硫酸钠浓度在1~5%范围内,2号水槽后与3号膜前的膜内间苯二胺残留量均在5mg/m2以下,实施例16~17的反渗透膜片初始脱盐率均在99.6%以上,初始渗透通量均在50.0l/(m2·
h)以上。然而,对比例6中由于4号水槽亚硫酸钠的浓度低于1%,反渗透膜内引入的磺酸基数量不够,导致反渗透膜的初始通量低于50.0l/(m2·
h),初始脱盐率低于99.6%。
[0186]
本领域技术人员可以理解,在本说明书的教导之下,可以对本发明做出一些修改或调整,这些修改或调整也应该在本发明权利要求所限定的范围之内。
[0187]
本发明的实施例仅需要在现有生产线的工艺不变的前提下,通过添加添加剂的方式,即可在保持高渗透通量和高脱盐率的前提下,提高反渗透膜的耐氧化性能和脱盐层结合强度,在产业上具有较好的可利用性。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1