Si-TiO2/g-C3N4三元复合光催化材料及其制备方法
si-tio2/g-c3n4三元复合光催化材料及其制备方法
技术领域
1.本发明涉及复合催化剂制备技术领域。具体地说是si-tio2/g-c3n4三元复合光催化材料及其制备方法。
背景技术:
2.随着我国工业化、城镇化的快速发展,水资源开发利用强度大,排放污染物的数量和种类增多,尤其是有毒、难降解的有机废水,对环境的危害越来越严重,大部分城市水污染严重。在水污染中,含有大量有机污染物的工业废水对人类的危害最大,这类废水主要包括简单的芳香族化合物、脂肪酸和芳香酸、脂肪醇等,染料、农药等有机废水也包含在内。目前,常用的处理水污染方法有物理处理、化学处理、生物处理和半导体光催化技术等手段。
3.半导体光催化技术作为一种新型环保技术,被认为是解决能源短缺和环境污染问题的有效方法之一。tio2因具有价格低、无毒无害、无二次污染、稳定性好、降解完全等优点成为半导体剂的研究热点。然而,二氧化钛光催化剂存在一个瓶颈,例如,它对紫外光的响应很好。此外,光生载流子和空穴容易结合,导致光催化活性和降解效率降低。研究表明,光催化效率取决于光催化材料的光响应能力和光生电子空穴的复合速率。为了提高光催化效率,必须扩大催化剂的光吸收范围,抑制光生电子空穴的复合。研究表明tio2的改性方法有贵金属沉积、半导体复合、染料光敏化、离子掺杂等。这些方法可以拓宽tio2颗粒裸露的比表面积和提高其可见光的响应范围,提高光生载体的分离和转移效率,进而使光催化活性提高。
4.掺杂因具有改性方法简单、改性效果好、可以实现半导体光催化剂的可见光性等优点被广泛用于改性研究。刘楠楠等用溶胶-凝胶法制备出fe/n、cu/n共掺杂和fe、cu、n单离子掺杂的tio2薄膜,在可见光下,cu/n掺杂tio2薄膜对盐酸四环素的降解率达到69%,对比tio2的光催化活性有明显的升高。zhao等采用简单水热法成功制备了ti
3+
、n共掺杂改性tio2复合材料,实验结果表明,共掺杂催化剂的光催化活性明显高于本体。王菲凤以氯化铵作为氮源,采用溶胶-凝胶法制备了n-tio2纳米材料,光催化降解水中的腐殖酸,当8%n-tio2的投加量为1mg/l时,其降解效率达80.32%。而不同禁带宽度的半导体材料以面对面的形式接触通过耦合构筑异质结进行复合,能够有效实现可见光响应特性。安涛等采用水热法制备的tio2/bi2wo6复合材料,光催化6min后对罗丹明b的降解率达99.3%,光催化30min后对亚甲基蓝的降解率达99.7%,光催化15min后对tc-hcl的降解率达87.7%。
5.目前,半导体光催化材料tio2只对紫外光有良好的响应,加之光激发后光生电子-空穴对容易复合,降低了其光催化活性。g-c3n4因存在比表面积较小,可见光响应范围有限,导致其光催化活性欠佳。
技术实现要素:
6.为此,本发明所要解决的技术问题在于提供一种降解速率快、降解率高的si-tio2/g-c3n4三元复合光催化材料及其制备方法,以解决当前半导体光催化材料tio2光生电
子-空穴对容易复合以及g-c3n4可见光响应范围有限等原因造成的光催化活性不理想等问题。
7.为解决上述技术问题,本发明提供如下技术方案:
8.si-tio2/g-c3n4三元复合光催化材料的制备方法,包括如下步骤:
9.(1)g-c3n4材料的制备:将三聚氰胺放入石英舟中,并将石英舟置于管式炉中烧结,将烧结后得到的固体进行粉碎,即制备得到g-c3n4材料;
10.(2)si-tio2材料的制备:将钛酸四丁酯与无水乙醇混合搅拌,得到溶液a;将无水乙醇、水、十六烷基三甲基溴化铵、正硅酸四乙酯和冰乙酸混合后搅拌均匀得到溶液b;将溶液a加入到溶液b中,搅拌均匀后得到混合溶液;先对混合溶液进行超声分散,然后将混合溶液置于鼓风烘箱中进行水热反应,反应结束后自然冷却至室温,将反应体系中的固体物质依次用水和无水乙醇洗涤,并进行干燥,得到块状固体;将块状固体进行研磨煅烧后即制备得到si-tio2材料;
11.(3)si-tio2/g-c3n4三元复合光催化材料的制备:将g-c3n4材料和si-tio2材料进行混合研磨,研磨均匀后,转移至石英舟中,并将石英舟置于管式炉中烧结,即制备得到si-tio2/g-c3n4三元复合光催化材料。
12.上述si-tio2/g-c3n4三元复合光催化材料的制备方法,在步骤(1)中,烧结时,管式炉以2~6℃/min的升温速率升温至520~560℃,并保温1~5h。【当升温速率过低或过高时,制备得到的光催化材料性能均较差,而当管式炉以5℃/min的升温速率升温至550℃时,制备得到的g-c3n4能够和si-tio2更高效地构建形成异质结,且得到的异质结稳定性更好,从而得到复合效果较好的si-tio2/g-c3n4三元复合光催化材料。另外,烧结温度过高或过低均会影响所制备g-c3n4材料的形貌结构及含碳量,从而不利于制备得到光催化活性较高的三元复合光催化材料,且当温度过高时,对材料的碳含量以及形貌结构影响尤为明显】。
13.上述si-tio2/g-c3n4三元复合光催化材料的制备方法,在步骤(2)中,配置溶液a时,钛酸四丁酯与乙醇的体积比为1:(1~3),搅拌时间为10~40min。
14.上述si-tio2/g-c3n4三元复合光催化材料的制备方法,在步骤(2)中,配置溶液b时,依次将无水乙醇、水、十六烷基三甲基溴化铵、正硅酸四乙酯和冰乙酸倒入容器中搅拌10~40min;无水乙醇、水、正硅酸四乙酯和冰乙酸的体积比为(15~18):(1~4):(0.1~0.3):(6~9),十六烷基三甲基溴化铵与水的质量之比为1:30~60。【在配置溶液b时,无水乙醇、水、十六烷基三甲基溴化铵、正硅酸四乙酯和冰乙酸要按照上述顺序进行混合,这种混合顺序可以使上述5种组分更容易混合均匀,有利于水热反应的进行,且可以使制备的三元复合光催化材料具有更好的形貌结构;若不按照上述顺序进行混合,则不论怎么调整其它工艺参数,都难以得到较理想的形貌结构;这是因为,如果不按照上述顺序混合,则正硅酸四乙酯更易与水发生反应形成絮状物,絮状物的存在不仅不利于水热反应的进行,而且影响三元复合光催化材料的形貌结构】;【在溶液a和溶液b进行水热反应时,无水乙醇在水热反应中提供压力,十六烷基三甲基溴化铵ctab在反应中作为催化剂,正硅酸四乙酯提供si原,可以与钛酸四丁酯提供的ti源形成si-tio2,冰乙酸和水作为形貌引导剂;本发明发现当无水乙醇、水、正硅酸四乙酯和冰乙酸的体积比为17.5:2.5:0.153:7.5、钛酸四丁酯与乙醇的体积比为1:2,且溶液a与溶液b的体积之比为1:2时,si离子能够更好地嵌入到tio2骨架中,得到较理想的si-tio2材料】。
15.上述si-tio2/g-c3n4三元复合光催化材料的制备方法,在步骤(2)中,水热反应的温度为80~160℃,水热反应的时间为6~14h;si-tio2材料中,si的质量分数为9~17wt%。
16.上述si-tio2/g-c3n4三元复合光催化材料的制备方法,在步骤(3)中,g-c3n4材料和si-tio2材料的质量之比为(1:3)~(3:1);烧结条件为:460~520℃煅烧2~5h,然后自然冷却至室温。
17.上述si-tio2/g-c3n4三元复合光催化材料的制备方法,在步骤(2)中,干燥温度为75~90℃,干燥时间为6~9h;块状固体进行研磨煅烧时的煅烧温度为460~520℃,煅烧时间为2~5h。
18.上述si-tio2/g-c3n4三元复合光催化材料的制备方法,在步骤(2)中,溶液a和溶液b混合后搅拌10~30min,超声分散25~40min,超声功率为400w;对所述固体物质洗涤时,先用水将固体物质洗涤2~3次,再用无水乙醇将固体物质洗涤2~3次。
19.上述si-tio2/g-c3n4三元复合光催化材料的制备方法,在步骤(1)中,烧结时,管式炉以5℃/min的升温速率升温至550℃,并保温3h;
20.在步骤(2)中,配置溶液a时,钛酸四丁酯与乙醇的体积比为1:2,搅拌时间为30min;配置溶液b时,依次将无水乙醇、水、十六烷基三甲基溴化铵、正硅酸四乙酯和冰乙酸倒入容器中搅拌30min;无水乙醇、水、正硅酸四乙酯和冰乙酸的体积比为17.5:2.5:0.153:7.5,十六烷基三甲基溴化铵与水的质量之比为1:50;水热反应的温度为140℃,水热反应的时间为10h;si-tio2材料中,si的质量分数为15wt%;
21.在步骤(2)中,干燥温度为80℃,干燥时间为8h;块状固体进行研磨煅烧时的煅烧温度为500℃,煅烧时间为3h;溶液a和溶液b混合后搅拌15min,超声分散30min,超声功率为400w;对所述固体物质洗涤时,先用水将固体物质洗涤2~3次,再用无水乙醇将固体物质洗涤2~3次;
22.在步骤(3)中,g-c3n4材料和si-tio2材料的质量之比为1:1;烧结条件为:500℃煅烧3h,然后自然冷却至室温。
23.si-tio2/g-c3n4三元复合光催化材料,由上述si-tio2/g-c3n4三元复合光催化材料的制备方法制备得到。
24.本发明的技术方案取得了如下有益的技术效果:
25.(1)本发明提供了一种si-tio2/g-c3n4三元复合光催化材料及其制备方法,采用本发明的方法制备得到的si-tio2/g-c3n4三元复合光催化材料在300w氙灯辐照90min时,对亚甲基蓝的降解率可达90.85%,降解速率达到0.03266min-1
。
26.(2)本发明采用高温煅烧法制备得到g-c3n4材料,采用水热反应法制备得到si-tio2二元复合材料,然后再采用高温煅烧法将g-c3n4材料和si-tio2二元复合材料复合生成si-tio2/g-c3n4三元复合光催化材料。该三元复合材料相比一元和二元光催化材料,在分解亚甲基蓝时具有降解速率快,降解率高的优点。这主要是由于本发明通过控制水热反应的时间和温度、si的掺杂量以及g-c3n4材料与si-tio2的质量比等工艺参数,制备得到的三元复合光催化材料收缩了tio2的禁带宽度,降低了光生电子-空穴对的复合几率,将吸收光范围延伸至可见光区域,从而提升了复合光催化材料的光催化效率。
附图说明
27.图1本发明实施例中光催化降解装置图;
28.图2本发明实施例中不同光催化剂的xrd图(10~80
°
);
29.图3本发明实施例中不同光催化剂的xrd图(放大至20~30
°
);
30.图4本发明实施例中不同光催化剂的ft-ir图;
31.图5本发明实施例中不同光催化剂光催化降解性能对照图;
32.图6本发明实施例中不同光催化剂在降解过程中的一级动力学拟合曲线图;
33.图7本发明实施例中不同水热时间制备的复合催化剂降解性能对照图;
34.图8本发明实施例中不同水热时间制备的复合催化剂在降解过程的一级动力学拟合曲线图;
35.图9本发明实施例中不同水热温度制备的复合催化剂降解性能对照图;
36.图10本发明实施例中不同水热温度制备的复合催化剂在光降解过程中的化学一级动力学拟合曲线;
37.图11本发明实施例中不同si掺杂制备的复合催化剂降解性能对照图;
38.图12本发明实施例中不同si掺杂制备的复合催化剂在光催化过程中的一级动力学拟合曲线;
39.图13本发明实施例中不同g-c3n4质量比制备的复合催化剂光催化降解对比图;
40.图14本发明实施例中不同g-c3n4质量比制备的复合催化剂在降解过程的化学一级动力学拟合曲线;
41.图15本发明实施例中三元复合光催化剂光催化机理降解图。
具体实施方式
42.1.si-tio2/g-c3n4三元复合光催化材料的制备方法,包括如下步骤:
43.(1)g-c3n4材料的制备:将三聚氰胺放入石英舟中,并将石英舟置于管式炉中烧结,将烧结后得到的固体进行粉碎,即制备得到g-c3n4材料;
44.(2)si-tio2材料的制备:将钛酸四丁酯与无水乙醇混合搅拌,得到溶液a;将无水乙醇、水、十六烷基三甲基溴化铵、正硅酸四乙酯和冰乙酸混合后搅拌均匀得到溶液b;将溶液a加入到溶液b中,搅拌均匀后得到混合溶液;先对混合溶液进行超声分散,然后将混合溶液置于鼓风烘箱中进行水热反应,反应结束后自然冷却至室温,将反应体系中的固体物质依次用水和无水乙醇洗涤,并进行干燥,得到块状固体;将块状固体进行研磨煅烧后即制备得到si-tio2材料;
45.(3)si-tio2/g-c3n4三元复合光催化材料的制备:将g-c3n4材料和si-tio2材料进行混合研磨,研磨均匀后,转移至石英舟中,并将石英舟置于管式炉中烧结,即制备得到si-tio2/g-c3n4三元复合光催化材料。
46.在步骤(1)中,烧结时,管式炉以5℃/min的升温速率升温至550℃,并保温3h。
47.在步骤(2)中,配置溶液a时,钛酸四丁酯与乙醇的体积比为1:2,搅拌时间为30min;配置溶液b时,依次将无水乙醇、水、十六烷基三甲基溴化铵、正硅酸四乙酯和冰乙酸倒入容器中搅拌30min;无水乙醇、水、正硅酸四乙酯和冰乙酸的体积比为17.5:2.5:0.153:7.5,十六烷基三甲基溴化铵与水的质量之比为1:50;水热反应的温度为140℃,水热反应的
时间为10h;干燥温度为80℃,干燥时间为8h;块状固体进行研磨煅烧时的煅烧温度为500℃,煅烧时间为3h;溶液a和溶液b混合后搅拌15min,超声分散30min,超声功率为400w;在对固体物质洗涤时,先用水将固体物质洗涤3次,再用无水乙醇将固体物质洗涤3次。si-tio2材料中,si的掺杂比为15wt%。
48.在步骤(3)中,g-c3n4材料和si-tio2材料的质量之比为1:1;烧结条件为:500℃煅烧3h,然后自然冷却至室温。
49.本实施例采用上述方法制备得到了si-tio2/g-c3n4三元复合光催化材料:主要以ctab(十六烷基三甲基溴化铵)表面活性剂作为软模板,采用水热法制备si-tio2光催化材料,利用高温煅烧法制备类石墨相氮化碳,将其与si-tio2通过高温煅烧复合得到si-tio2/g-c3n4三元复合光催化材料。本实施例的具体实验方法及实验过程详细如下。
50.2实验部分
51.2.1试剂与仪器
52.本实施例所用的主要化学试剂及实验仪器分别见表2-1和表2-2。
53.表2-1主要化学试剂
[0054][0055]
表2-2主要实验仪器
[0056][0057]
2.2实验方案
[0058]
2.2.1 g-c3n4材料的制备
[0059]
操作电子分析天平取三聚氰胺(5g)为原料放置于石英舟,然后将石英舟放置于管
式炉中,温度上升到550℃,加热速率为5℃/min,在550℃下持续3小时。待冷却后,将产物从石英舟中取出,得到淡黄色块状固体即为石墨相氮化碳(g-c3n4)样品,将块状固体用玛瑙研钵研磨均匀成细粉状装瓶待用。
[0060]
2.2.2 tio2材料的制备
[0061]
量取5ml钛酸四丁酯加入10ml无水乙醇搅拌30min配置a溶液;移取17.5ml无水乙醇、2.5ml水和7.5ml冰乙酸,按照先后顺序加入烧杯中搅拌30min配置b溶液;将搅拌好的b溶液缓慢加入a溶液中,边加入边搅拌,混合均匀后继续搅拌15min。待搅拌完成后,将混合后的溶液超声分散30min,待超声结束后,将超声分散的溶液转置反应釜中进行水热反应,水热反应的温度为140℃,水热反应时间为10h,反应结束后自然冷却到室温,将反应釜中的上清液清理后,剩余饼状固体转置于离心管中,用去离子水和无水乙醇洗涤。将所得产物转置干燥箱中在80℃中干燥8h,将干燥后的块状物进行研磨,研磨后置于管式炉中以5℃/min的速率升温至500℃,保温3h,得到粉末状固体,即为tio2材料。
[0062]
2.2.3 tio2/g-c3n4材料的制备
[0063]
将制备好的tio2和g-c3n4按照一定的质量比混合并研磨均匀后,在管式炉中以5℃/min的速率升温至500℃,保温3h,待自然冷却后,得到淡黄色粉末,即为tio2/g-c3n4材料。
[0064]
2.2.4 si-tio2材料的制备
[0065]
量取5ml钛酸四丁酯加入10ml无水乙醇搅拌30min配置a溶液;移取17.5ml无水乙醇、2.5ml水、0.05g ctab、0.153ml正硅酸四乙酯、7.5ml冰乙酸,按先后顺序加入烧杯中搅拌30min配置b溶液,将搅拌好的a溶液缓缓加入b溶液中,混合均匀后搅拌15min。待搅拌完成后,将混合后的溶液超声分散30min。待超声结束后,将溶液转置200ml反应釜中,在鼓风烘箱中设置一定温度和时间进行水热反应,反应结束后自然冷却到室温,将反应釜中的上清液清理后,剩余饼状固体转置于离心管中,用去离子水和无水乙醇洗涤3次。将所得产物转置干燥箱中在80℃中干燥8h,将干燥后的块状物进行研磨煅烧,煅烧时,以5℃/min的速率升温至500℃,保温3h,得到白色粉末状固体,即为si掺比为15wt%的si-tio2光催化材料,表示为15%si-tio2;按照同样的方法,制备出9%si-tio2、11%si-tio2、13%si-tio2、15%si-tio2和17%si-tio2。
[0066]
2.2.5 si-tio2/g-c3n4复合材料的制备
[0067]
称取一定质量的(x)si-tio2和g-c3n4材料于研钵中进行研磨至均匀后,将样品放入石英舟中,在500℃温度下煅烧3h,自然冷却后得到淡黄色的粉末状固体,即为(x)si-tio2/g-c3n4复合型光催化剂。(x)si-tio2代表si掺比为xwt%的si-tio2光催化材料。
[0068]
本实施例通过调整x大小或(x)si-tio2和g-c3n4材料的质量之比,按照同样的方法制备得到不同的(x)si-tio2/g-c3n4材料。
[0069]
2.3 si-tio2/g-c3n4三元复合光催化材料的结构表征
[0070]
xrd表征采用cukα为辐射源,在10~80
°
扫描范围内,对制备得到的si-tio2/g-c3n4三元复合光催化材料样品的晶相结构进行扫描;ft-ir表征以0.0001cm-1
的精度,在3500-500cm-1
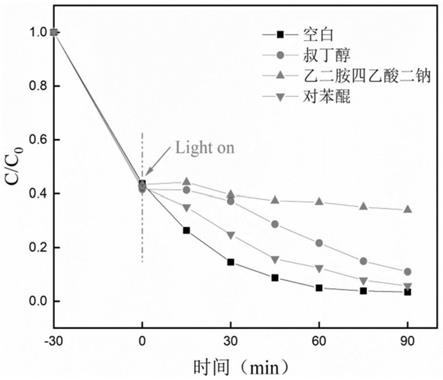
的扫描范围内研究光催化材料的特征峰和结构的特点。
[0071]
2.4光催化降解实验
[0072]
采用如图1的光催化降解装置,用于样品对污染物的降解实验。实验流程如下:用
玻璃棒搅拌,将20mg的亚甲基蓝(mb)溶于盛有适当蒸馏水的烧杯中,用玻璃棒引流将溶液和润洗液转置于1000ml容量瓶内,加水至刻度线定容,配置得到20mg/l的亚甲基蓝水溶液。量取50ml亚甲基蓝水溶液于预先添加了25mg三元复合光催化剂(si-tio2/g-c3n4)的石英管中;纯光催化剂(tio2、g-c3n4材料)和二元复合光催化剂(tio2/g-c3n4、si-tio2光催化材料)做相同处理,用以作对比。在石英管中放入磁子置于黑暗环境中,用磁子搅拌器搅拌,作暗反应处理,使光催化材料样品和亚甲基蓝水溶液之间达到吸附-解析平衡状态。暗反应30min后,用移液枪取4ml样品放置于已编号的取样盘中,其他四组光催化剂做相同处理。将五组光催化剂(分别为:tio2、g-c3n4材料、tio2/g-c3n4、si-tio2光催化材料和si-tio2/g-c3n4三元复合光催化剂)放在300w氙灯下,继续磁子搅拌,做光催化实验,每15min取4ml样品放置于取样盘中,光反应90min。光催化降解实验结束后,将样品在离心机中进行离心操作,以使漂浮的小颗粒沉降;用滴管小心吸取上层溶液至比色皿中进行吸光度数值的测试。
[0073]
2.5标准曲线的绘制
[0074]
标准曲线的绘制有利于体现光催化剂的吸光度和染料浓度随光催化反应的变化规律。详细实验步骤如下:按照标准溶液配置法,分别调配5mg/l、10mg/l、15mg/l、20mg/l、25mg/l的亚甲基蓝水溶液。分别将配置好的五组亚甲基蓝水溶液取4ml于标号的取样盘中,使用离心机在适当的转速下对所取样液离心。采用可见分光光度计测不同浓度亚甲基蓝水溶液的吸光度,因吸光度数值会出现波动,故取三次数值求其平均值。用散点图在绘图软件中进行绘制后,对其进行数据拟合,即为标准曲线。
[0075]
2.6光催化机理实验
[0076]
在光的激发下,光催化材料会产生多种活性物质,其中主要包括空穴和自由基,为探讨活性物质在光催化剂光反应降解染料中的贡献大小,需进行以下实验操作:取4根石英管进行标号,记为a、b、c、d,洗涤干燥后待用。用电子分析天平分别称取25mg三元复合材料si-tio2/g-c3n4于石英管中,放入大小适中的磁子,加入亚甲基蓝溶液50ml,将石英管转置于光催化装置,用磁子搅拌器搅拌,在黑暗环境中反应30min后取样。在进行光反应之前,a组为空白对照,b组中加入5mmol/l的叔丁醇,c组中加入5mmol/l的乙二胺四乙酸二钠,d组中加入0.5mmol/l的对苯醌进行光催化实验。光催化实验完成后,在测吸光度之前将样品用离心机进行离心操作。
[0077]
3结果与讨论
[0078]
3.1 x射线衍射(xrd)分析
[0079]
为了进一步了解光催化材料si-tio2/g-c3n4复合对tio2和g-c3n4基本晶体结构的影响,分析了g-c3n4、tio2、tio2/g-c3n4、15%si-tio2和15%si-tio2/g-c3n4五种光催化材料xrd图谱,其中,制备tio2/g-c3n4时,tio2与g-c3n4的质量之比为1:1;制备15%si-tio2以及15%si-tio2/g-c3n4中的15%si-tio2时,水热反应的温度为140℃,水热反应时间为10h;制备15%si-tio2/g-c3n4时,15%si-tio2与g-c3n4的质量之比为1:1。从图2和图3可以看出,制备的g-c3n4在12.9
°
和27.3
°
处有特征峰,分别对应(100)晶面和(002)晶面,与标准卡g-c3n4材料的特征峰相对应,表明所制备的物质为g-c3n4。样品tio2在25.4
°
、37.9
°
、48.1
°
、54.2
°
、55.1
°
、63.7
°
、68.9
°
处有七个特征峰,分别对应(101)、(103)、(004)、(112)、(200)、(105)、(211)晶面,分别与锐钛矿相tio2材料得特征峰相符合,并不含金红石相和板钛矿相特征峰,表明制备的tio2为锐钛矿相,且具有较高的纯度和结晶度。在tio2/g-c3n4、15%si-tio2、
15%si-tio2/g-c3n4样品的图谱中,它们的特征峰均与锐钛矿相二氧化钛的特征峰保持一致,大体上偏差不大,表明复合效果良好,然而观察图谱会发现,复合材料的特征峰强度会在一定程度上减弱,其中三元复合材料15%si-tio2/g-c3n4的特征峰强度最弱。另外,与g-c3n4构建异质结的复合材料,其衍射峰的角度相对会有些许偏移。
[0080]
通过掺杂和构建异质结制备的光催化材料15%si-tio2/g-c3n4在25.4
°
处的特征峰减弱,一方面是因为si离子嵌入tio2骨架中,si-tio2和g-c3n4以面对面形式成功构建异质结所导致的;另一方面,是由于材料在高温条件下的聚合反应影响所影响的。复合材料的衍射角有些许偏移,可能受半导体之间的相互作用产生的作用力导致tio2的晶格在热沉积过程中的收缩和变形所影响。而在三元复合材料图谱中并未出现石墨相氮化碳的特征峰,可能是由于材料是经过高温煅烧复合制备而来,在煅烧过程中,复合材料中的g-c3n4在500℃下轻微分解。
[0081]
3.2傅里叶红外光谱(ft-ir)分析
[0082]
如图4所示采用ft-ir光谱分析光催化材料的官能团归属,共分析了g-c3n4、tio2、tio2/g-c3n4、15%si-tio2和15%si-tio2/g-c3n4五种光催化材料的红外光谱,其中,制备tio2/g-c3n4时,tio2与g-c3n4的质量之比为1:1;制备15%si-tio2以及15%si-tio2/g-c3n4中的15%si-tio2时,水热反应的温度为140℃,水热反应时间为10h;制备15%si-tio2/g-c3n4时,15%si-tio2与g-c3n4的质量之比为1:1;在测量时,应将待测的光催化样品材料与kbr以体积比1:100充分研磨均匀、压片,结果如图所示。从图中可以看出,在1623cm-1
处的吸收峰为吸附在光催化剂表面的水分子的弯曲振动;805cm-1
和1200-1600cm-1
的范围存在振动峰,这两组振动峰分别与碳氮三嗪单元的弯曲振动和芳香碳氮杂环伸缩振动所引起的振动峰范围相对照,表明样品为g-c3n4。而在与g-c3n4构建异质结的催化剂样品中在相同区域也包含相似的振动峰。在复合材料ft-ir光谱中,在910cm-1
和1048cm-1
处出现的弱吸收带分别是ti-o-si和si-o-si的伸缩振动;在其他位置出现的吸收峰,分别与tio2、g-c3n4相对应,表明复合效果良好。
[0083]
3.3光催化活性结果与讨论
[0084]
将通过光催化实验得到的吸光度数值,通过标准曲线换算成相对应的浓度。将亚甲基蓝(mb)的初始浓度记为c0,c为t时刻取样的mb浓度,以c/c0为纵坐标,反应时间t作折线图,该折线图能够直观反映出体系中三元复合光催化材料15%si-tio2/g-c3n4对模拟污染物mb的降解情况。其中亚甲基蓝溶液的降解率计算公式为:
[0085][0086]
3.3.1不同光催化材料光催化性能的研究
[0087]
将制备的复合材料和相同实验条件下制备的tio2、g-c3n4、5%si-tio2、tio2/g-c3n4和5%si-tio2/g-c3n4分别称取25mg,放入装有50ml亚甲基蓝的石英管中,用磁子搅拌,暗反应30min后取4ml样品后,剩余溶液在300w氙灯辐照下进行光催化反应,每15min取一次样,光反应90min,光反应结束后,将样品离心进行吸光度检测。此处,制备5%si-tio2以及5%si-tio2/g-c3n4中的5%si-tio2时,水热反应温度为140℃,水热反应时间为12h;制备tio2/g-c3n4时,tio2与g-c3n4的质量之比为1:1;制备5%si-tio2/g-c3n4时,5%si-tio2与g-c3n4的质量之比为1:1。
[0088]
图5和图6为上述不同光催化剂在相同实验条件下降解亚甲基蓝的折线图,从图中可以看出,三元复合材料5%si-tio2/g-c3n4对亚甲基蓝的降解效果要明显优于tio2、g-c3n4、tio2/g-c3n4、5%si-tio2光催化剂,其在300w氙灯辐照90min,降解率达到85.7%,降解速率为0.02438min-1
。其降解速率分别为tio2的9.34倍、g-c3n4的9.67倍、tio2/g-c3n4的4.31倍、5%si-tio2的1.95倍。
[0089]
在三元复合材料的制备过程中,涉及到很多影响因素。为进一步优化复合催化剂的光催化降解性能,对其制备条件进行了探究。在实验中,采用控制变量法,对水热时间、水热温度、si、g-c3n4在复合材料中的不同比例等因素通过平行实验来探究对三元复合材料光催化活性的影响。
[0090]
3.3.2水热时间对复合材料光催化性能的影响
[0091]
在研究不同水热时间对光催化性能的实验中,将a、b溶液混合搅拌超声后,转置于200ml反应釜中,将水热温度定为140℃进行水热反应实验,分别在鼓风烘箱中反应6、8、10、12、14h反应结束后依次按照洗涤、离心、干燥和高温煅烧等步骤来制备复合材料,并对其光催化性进行研究。在制备光催化剂时,硅的掺比为5%,5%si-tio2与g-c3n4的质量之比为1:1。
[0092]
图7和图8为不同水热反应时间制备的三元复合材料5%si-tio2/g-c3n4降解亚甲基蓝溶液的折线图,光催化实验表明,当水热时间低于10h,随着水热时间的增加,三元复合材料5%si-tio2/g-c3n4的光催化活性也会呈梯度提高,当水热反应时间为10h时,光催化剂在300w氙灯辐照90min,降解率为88.51%,降解速率为0.03928min-1
光催化性能最好。但随着反应时间的变长,当水热时间高于10h时,复合材料对亚甲基蓝的光催化活性呈现梯度下降的趋势,即过长的水热反应时间会降低三元复合材料的光催化性能,这可能是由于过长的水热时间导致光催化材料的颗粒直径增加、比表面积减小的结果。而在10h时的光催化性能最佳,一方面,这可能反应时间可以改变复合材料的晶体大小,更有利于增强反应物和产物的吸附和脱附;另一方面,在这个温度区间,光催化材料具有更高的比表面积、小的晶粒尺寸和较好的晶化。由于水热时间过短,复合材料没有形成具有较高光催化活性的晶体和形貌,导致光催化活性偏差。
[0093]
3.3.3水热温度对复合材料光催化性能的影响
[0094]
在研究不同水热温度对光催化性能的实验中,将溶液a和溶液b混合搅拌超声后,倒入反应釜中,在80、100、120、140、160℃反应10h,在反应结束后依次按照上述实验步骤制备复合材料,并通过做光催化实验对这五组不同水热反应温度制备的三元复合材料进行研究。在制备光催化剂时,硅的掺比为5%,5%si-tio2与g-c3n4的质量之比为1:1。
[0095]
图9和图10为不同水热温度下制备的光催化材料5%si-tio2/g-c3n4降解亚甲基蓝溶液的折线图。从图中可以直观明显的看到,当水热温度低于140℃时,随着反应温度的升高,复合材料的光催化活性即对亚甲基蓝的降解率呈梯度增加,当水热反应时间为140℃时,光催化活性和降解率达到峰值,用300w氙灯辐照90min,复合材料对亚甲基蓝(mb)的降解率可达88.63%,降解速率为0.02825min-1
。而当温度超过140℃时,复合材料的光催化活性继而会下降。这可能是因为在恰当的温度区间,三元复合材料5%si-tio2/g-c3n4具有完整的晶形、恰当的晶粒粒径和比表面积,从而降低了光生电子和空穴的复合概率,导致单位面积上发生反应的机率就越大。而当水热反应温度较低时,虽然光催化材料有更大的比表
面积和更小的晶粒粒径,但由于其晶型不完整,光催化活性反而下降。随着水热反应温度的升高,复合材料的晶粒可能会变大,这一情况与xrd图谱相结合分析可得出水热温度的升高会导致三元复合材料晶粒的长大和晶化的增强。另外,在3.3.2”中,5%si-tio2/g-c3n4三元复合材料制备时的水热反应时间也是10h,反应温度同样为140℃,但其对亚甲基蓝的降解率为88.51%,降解速率为0.03928min-1
;而在本部分实验中,5%si-tio2/g-c3n4三元复合材料对亚甲基蓝的降解率为88.63%,降解速率为0.02825min-1
;由此可见,相同工艺参数条件下制备的三元复合材料,对亚甲基蓝的降解率在不同次实验中基本相同,但降解速率却有很大差别,这主要是因为降解速率是用来表示催化剂在单位时间内对模拟污染物的降解效果,在实验过程中,可能和磁子转速和温度有关,而降解率则可以更好的评价材料的光催化性能。
[0096]
3.3.4 si掺杂比例对复合材料光催化性能的影响
[0097]
在研究si元素在复合材料中的不同比例对复合材料光催化活性影响的实验中,通过改变si元素的掺杂量,按照实验流程,一共制得五组(x)si-tio2/g-c3n4材料,其中x取(9wt%、11wt%、13wt%、15wt%、17wt%),以确定光催化剂(x)si-tio2/g-c3n4的最佳掺杂比例。在制备不同si掺比的(x)si-tio2/g-c3n4时,先制备(x)si-tio2,制备(x)si-tio2时,水热反应的温度是140℃,水热反应的时间是10h,在制备(x)si-tio2/g-c3n4时,(x)si-tio2与g-c3n4的质量之比1:1;如图11和图12所示为光催化剂对亚甲基蓝的降解效果和降解过程中的以及动力学拟合曲线图。
[0098]
从图11和图12中可以直观的看出,不同的si元素掺杂量会导致复合材料吸附和降解性能的不同。在si元素掺杂比例小于15wt%时,三元复合材料si-tio2/g-c3n4的吸附和光催化活性会随着掺杂si元素比例的增加而变强,当si元素掺杂比例为15wt%时,复合材料的吸附和降解性能达到峰值,继续加大si元素掺杂含量,则会发现,其吸附和光催化活性继而下降,表明当si元素掺杂含量为15wt%时,复合材料具有更高的吸附和光催化性能。这可能是因为si
4+
的掺杂在复合催化剂表面形成ti-o-si结构,在所对应的禁带区间实现的能级的跃迁,产生可见光响应,实现和提高催化剂表面光生载流子的直接分离,提高了光催化活性;另一方面,si
4+
的掺杂使复合材料具有较小的平均粒径和较大的比表面积,导致光生载流电子更容易的迁移到粒子表面,从而在催化剂表面提供了更多活性物种(羟基),对亚甲基蓝的吸附和氧化能力增强,促进了光催化材料光催化活性的提高。而当si元素掺杂含量过高时,复合材料的光催化活性降低,可能是因为,过量的si堵塞了半导体材料的介孔结构,影响催化剂光激发后,自由电子的转移。
[0099]
3.3.5 g-c3n4复合比例对复合材料光催化性能的影响
[0100]
将用水热法(水热反应温度为140℃、水热反应时间为10h)制备好的15%si-tio2催化剂研磨成粉末状与通过以三聚氰胺为原料通过高温煅烧法制备的g-c3n4以质量比3:1、2:1、1:1、1:2、1:3的比例研磨混合均与后,高温煅烧制备复合材料,并进行光催化实验,探究其光催化性能。
[0101]
将五组不同g-c3n4复合比例的三元复合材料进行光催化实验,经过一定时间的暗反应和光催化反应后,结果如图13和图14所示,当质量比15%si-tio2:g-c3n4为1:1时,三元复合材料的光催化活性最好,用300w氙灯辐照90min,复合材料对亚甲基蓝(mb)的降解率可达90.85%,降解速率为0.03266min-1
,明显要优于其他复合比例。这是由于在复合材料的制
备过程中,不是单纯将si-tio2、g-c3n4两个材料进行叠加,而是发生了一定的反应,从而产生了协同效应,大大增加了光催化活性。在高温煅烧法制备过程中,当g-c3n4含量过高时,si-tio2含量就会相对减少,g-c3n4表面形成的光生载流子无法高效地转移至tio2表面,大大降低光生电子-空穴对的分离速率,从而影响了光催化性能;当g-c3n4含量过低时,无法提供充足的光生载流子,降低了光催化活性。
[0102]
3.4三元复合材料的光催化机理
[0103]
在光催化降解体系中分别加入一定量的空穴、超氧自由基、羟基自由基的捕获剂乙二胺四乙酸二钠、对苯醌和叔丁醇,观察和分析三元复合材料15%si-tio2/g-c3n4对亚甲基蓝的降解情况随时间变化的情况,制备15%si-tio2/g-c3n4时,15%si-tio2与g-c3n4的质量之比为1:1,15%si-tio2制备时水热反应温度为140℃,水热反应时间为10h。如图15所示,在不添加任何捕获剂的空白实验中,三元复合材料的光催化降解性能最好,降解率为85.6%【本次实验与3.3.5中的实验结果有明显差别,这可能是因为,所制备的三元复合材料在放置过程中,自身结构会变得紧密,降低了其比表面积,从而导致降解率从90.85%下降到85.6%】,加入捕获剂乙二胺四乙酸二钠后,复合材料的光催化活性明显下降,降解率降为19.9%,空穴在其中被抑制,表明空穴h
+
在光催化降解实验中起到至关重要的作用。而加入的捕获剂苯醌和叔丁醇对超氧自由基、羟基自由基起到抑制作用,复合材料对染料的降解率分别为70.1%、79.8%,说明超氧自由基、羟基自由基对光催化降解染料也有共献,但没有空穴的贡献大。综上所述,光反应激发的三种活性物质对复合材料的光催化贡献,其贡献大小排序为h
+
》
·
oh》
·o2-。
[0104]
4结论
[0105]
由于半导体光催化材料tio2只对紫外光有良好的响应,加之光激发后光生电子-空穴对容易复合,降低了其光催化活性。g-c3n4因存在比表面积较小,可见光响应范围有限,导致其光催化活性欠佳。为弥补上述半导体材料的缺陷,本实施例采用水热法和高温煅烧法制备了si-tio2/g-c3n4三元复合光催化材料。并通过光催化实验和表征手段得出以下结论:
[0106]
(1)本实施例研究了水热反应时间、水热反应温度,以及si-tio2与g-c3n4在复合材料中的不同比例对三元复合材料降解亚甲基蓝光催化性能的影响,实验结果表明,在si掺杂量为15wt%时,140℃下水热反应10h,与g-c3n4在比例为1:1的条件下高温煅烧制备的复合材料具有最佳的吸附和光催化活性。
[0107]
(2)经过xrd、ft-ir光谱分析可知,离子掺杂和异质结的构建没有改变tio2和g-c3n4的基本结构,缩小了tio2的禁带宽度,降低了光生电子-空穴对的复合几率,使其吸收光范围延伸至可见光区域,提高了光的利用率,除此之外,光生载流子的高效迁移使复合材料表面具有更多的活性物种,提升了光催化效率。
[0108]
综上所述,本实施例通过水热法和高温煅烧法成功制备出具有优良性能的si-tio2/g-c3n4光催化剂,且当si掺杂量为15wt%时,140℃下水热反应10h,与g-c3n4在比例为1:1的条件下高温煅烧制备的复合材料具有最佳的吸附和光催化活性,在300w氙灯辐照90min时,对亚甲基蓝的降解率可达90.85%,降解速率达到0.03266min-1
,在降解水体有机污染物方面有积极意义和较好的发展前景。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1