一类基于VAPOL合成硒硫类化合物的合成方法与流程
一类基于vapol合成硒硫类化合物的合成方法
技术领域
1.本发明属于有机化学合成技术领域,具体涉及一类基于vapol合成硒硫类催化剂的合成方法。
背景技术:
2.类比于广为人知的binol配体在化学催化方向的广泛应用,vapol骨架分子是现有轴手性催化剂和配体的重要组成部分,同时也存在于具有生物活性的天然产物和药物分子中,也鉴于这种骨架的重要性,廉价、便捷的合成方法的开发是非常有必要的。为进一步丰富催化剂类型,催化更多的化学反应提供了更多选择。
3.william d. wulff是vapol结构的发现与拓展应用中至关重要的化学家,他首次于1993年提出了vapol的结构与binol结构对比,并就直接将vapol当作催化剂应用在手性催化反应中,并以优异的选择性拿到了产物。(j. am. chem. soc.1993, 115, 3814-3815.)并且于1996首次对vapol进行了合成和绝对构型的测定。(j. am. chem. soc.1996, 118, 3392-3405.)提出了两种合成vapol的方法,包括卡宾配合物与炔烃的反应和烯酮的[2+2]环加成反应。同年,他们将vapol衍生物作为催化剂催化了醛与烯丙基三丁基锡的不对称烯丙基化反应。2010年该课题组是比较早的开始合成了vapol的磷酸衍生物(synthesis. 2010, 21, 3670-3680.)和vapol的硼酸酯化物(j. am. chem. soc.2010, 132, 14669
–
14675.)并将其作为配体使用。近几年,vapol的磷酸胺衍生物作为配体催化剂的使用才慢慢发展起来。2021年,alois f
ü
rstner课题组利用vapol磷酰胺作为配体,在金属镍的存在下催化醛和二烯醇醚对映选择性合成相对应的产物;而vapol磷酸胺衍生的硒/硫化衍生物目前为止是没有人报道过的,化学结构上硒硫原子的引入在催化化学方面是有很大应用的,这方面已有很多化学家的报道验证了这一想法,同时这也丰富了不对称催化领域的催化剂库,为新的反应开发提供更多选择。
技术实现要素:
[0004]
本发明的目的就是为了克服上述现有技术存在的缺陷而提供一种反应条件温和,所使用原料便宜易制且基本无毒,可重复性好,底物适用范围广,经济性好的vapol磷酸胺衍生的硒/硫化物的合成方法。
[0005]
本发明为实现上述目的所采取的技术方案为:本发明的目的是提供一种手性vapol类催化剂,其结构式如下所示:其中,
r选自氢、甲基、乙基、丙基、异丙基、丁基、正丁基、异丁基、芳基、杂芳基。
[0006]
本发明中的手性vapol类催化剂对硫芳基参与的六元环手性内酯合成反应和/或轴手性含硫双苯基衍生物合成反应具有优良的催化活性。
[0007]
需要说明的是,本发明公开的手性vapol类催化剂为vapol类硒或硫化物,包括以下结构式:, ,,。
[0008]
本发明还公开了一种手性vapol类催化剂在催化硫芳基参与的六元环手性内酯合成反应和/或轴手性含硫双苯基衍生物合成反应中的用途。
[0009]
需要说明的是,作为优选的技术方案,本发明还公开了一种手性vapol类催化剂的合成方法,是将手性vapol与三氯化磷、三乙胺、烷基胺,在硒粉或硫粉以及溶剂的共同作用下,生成手性vapol类催化剂。
[0010]
需要说明的是,本发明公开的一种手性vapol类催化剂的合成方法为,将三氯化磷溶解于二氯甲烷中,冷却至低温-5~0℃后,并向其中逐滴滴加三乙胺,反应后将体系升至室温,并添加烷基胺,然后搅拌,在室温下向体系中加入手性vapol搅拌反应,室温条件下加入硒粉或硫粉,继续搅拌反应,后经减压蒸馏、柱色谱提纯,得到手性vapol类催化剂。
[0011]
本发明中手性vapol类催化剂的合成方法通过选择不同的烷基胺,首次实现了不同的手性vapol衍生硒/硫化物催化剂合成;且采用经济可购的vapol和廉价易得的烷基胺作为底物,一锅法制得手性的vapol衍生硒/硫化物催化剂,反应过程避免了复杂的合成的路线。
[0012]
需要说明的是,作为优选的技术方案,本发明公开的一种手性vapol类催化剂的合成方法中,烷基胺、手性vapol与硒粉的摩尔比为1:1:2~4。
[0013]
需要说明的是,作为优选的技术方案,本发明公开的一种手性vapol类催化剂的合成方法中,烷基胺、手性vapol与硫粉的摩尔比为1:1:2~4。
[0014]
需要说明的是,作为优选的技术方案,本发明公开的一种手性vapol类催化剂的合成方法中,烷基胺至少包括二甲胺、二乙基胺、二异丙胺、二正丁基胺、二异丁胺。
[0015]
与现有技术相比,本发明具有以下特点:1)本发明方法通过选择不同的烷基胺,首次实现了不同的手性vapol衍生硒/硫化物催化剂合成;2)本发明方法采用经济可购的vapol和廉价易得的烷基胺作为底物,一锅法制得手性的vapol衍生硒/硫化物催化剂,反应过程避免了复杂的合成的路线;3)本发明方法底物具有多样性,可以合成多种烷基取代基的vapol衍生硒/硫化物催化剂,其对硫芳基参与的六元环手性内酯合成反应和/或轴手性含硫双苯基衍生物合成反应具有优良的催化活性。
[0016]
因此,本发明是提供一种反应条件温和,所使用原料便宜易制且基本无毒,可重复性好,底物适用范围广,经济性好的vapol磷酸胺衍生的硒/硫化物的合成方法。
附图说明
[0017]
图1为化合物1a的1h核磁共振光谱;图2为化合物1a的
13
c核磁共振光谱;图3为化合物1b的1h核磁共振光谱;图4为化合物1b的
13
c核磁共振光谱;图5为化合物2a的1h核磁共振光谱;图6为化合物2a的
13
c核磁共振光谱;图7为化合物2b的1h核磁共振光谱;图8为化合物2b的
13
c核磁共振光谱;图9为化合物3a的1h核磁共振光谱;图10为化合物3a的
13
c核磁共振光谱;图11为化合物3b的1h核磁共振光谱;图12为化合物3b的
13
c核磁共振光谱;图13为化合物4a的1h核磁共振光谱;图14为化合物4a的
13
c核磁共振光谱;图15为化合物4b的1h核磁共振光谱;图16为化合物4b的
13
c核磁共振光谱;图17为化合物5c的1h核磁共振光谱;图18为化合物5c的
13
c核磁共振光谱;图19为化合物6c的1h核磁共振光谱;图20为化合物6c的
13
c核磁共振光谱;图21为化合物5c的消旋样品的hplc谱图;图22为化合物5c的手性样品的hplc谱图;图23为化合物6c的消旋样品的hplc谱图;图24为化合物6c的手性样品的hplc谱图。
具体实施方式
[0018]
针对现有技术中关于手性vapol类催化剂的合成研究较少的问题,本发明合成方法选择原料易得的烷基胺为原料,实现不同的手性vapol衍生硒/硫化物催化剂合成;且合成路线简单,能够得到收率较高的手性vapol类催化剂。
[0019]
下面结合具体实施方式对本发明进行进一步的详细描述,给出的实施例仅为了阐明本发明,而不是为了限制本发明的范围。以下提供的实施例可作为本技术领域普通技术人员进行进一步改进的指南,并不以任何方式构成对本发明的限制。如无特殊说明,本发明中的合成方法均为常规方法。本发明中所用的材料、试剂等,如无特殊说明,均可从商业途径得到。
[0020]
进一步需要说明的是,本发明公开的一种手性vapol类催化剂的合成方法为,将三氯化磷溶解于二氯甲烷中,冷却至低温0℃后,并向其中逐滴滴加三乙胺,反应5~20 min后将体系升至室温,并添加烷基胺,然后搅拌3~7h。在室温下向体系中加入手性vapol的衍生物搅拌反应10~15 h,室温条件下加入硒粉或硫粉,继续搅拌反应10~15 h。后经减压蒸馏、
柱色谱提纯,得到手性vapol类催化剂。
[0021]
具体合成路线为:。
[0022]
对于vapol衍生硒化物的催化应用1,提供一种制备硫芳基参与的六元环手性内酯的合成方法,具体合成路线如下:。
[0023]
对于vapol衍生硒化物的催化应用2,提供一种轴手性含硫双苯基衍生物的合成方法,具体合成路线如下:。
[0024]
本发明催化剂所应用的方法的设计思路是以易制的双苯基苯酚类底物和硫芳基试剂为原料,在催化剂和对氯苯磺酸的作用下,于低温下,氩气氛围中进行反应一段时间后,将体系升至一定温度,继续反应一段时间,制得轴手性含硫双苯基衍生物。
[0025]
下面通过实施例对本发明做进一步阐述,其目的仅在于更好理解本发明的内容。因此,本专利的保护范围并不限于这些实施例。
[0026]
本发明的实施方式中,化合物的氢核磁共振谱(1h nmr和
13
c nmr)由bruker avance iii hd 400测定,溶剂为氘代氯仿。化学位移(δ)以ppm为单位引用,以四甲基硅烷作为内标,多重性分别表示:s=单重态,d=双重态,t=三重态,q=四重态,m=多重态。
[0027]
本发明的合成方法中所述催化剂包括:
。
[0028]
以下结合具体实施方式对本发明的技术方案作进一步详细描述:实施例1:化合物1a的制备,包括:具体合成路线如下:。
[0029]
将0.13 mmol三氯化磷溶解于超干dcm(0.5 ml)中。将反应冷却至0
°
c,并向其中逐滴滴加0.65 mmol et3n。10 min后,将反应体系升至室温,并添加0.13 mmol二异丙胺,然后搅拌5 h。在室温下向体系中加入0.13 mmolvapol搅拌反应12 h后,室温条件下加入 0.39 mmol硒粉,搅拌反应12小时。反应完成后用硅藻土过滤,并在真空中浓缩、旋干。粗产物通过硅胶(1:60 etoac:石油醚)纯化,得到白色固体化合物1a,产率为50%。
[0030]1(400 mhz, cdcl3) δ 9.97-9.87 (m, 2h), 8.00-7.96 (m, 2h), 7.87-7.79 (m, 3h), 7.75-7.69 (m, 5h), 7.62 (d, j = 1.4 hz, 1h), 7.55 (d, j = 1.4 hz, 1h), 7.17
ꢀ–ꢀ
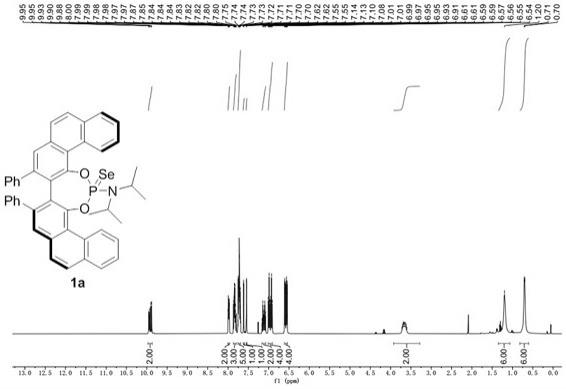
7.08 (m, 2h), 6.96 (dt, j = 23.6, 7.8 hz, 4h), 6.58 (ddd, j = 17.7, 8.3, 1.4 hz, 4h), 3.66 (ddt, j = 20.5, 13.1, 6.3 hz, 2h), 1.20 (s, 6h), 0.70 (d, j = 6.7 hz, 6h);如图1所示;
13
c nmr (101 mhz, cdcl3) δ 149.25, 149.09, 147.32, 147.24, 141.67, 141.65, 141.53, 141.51, 139.68, 139.63, 134.70, 134.69, 134.35, 134.33, 133.40, 133.13, 129.73, 129.59, 129.51, 129.29, 129.19, 129.12, 128.78, 128.74, 128.63, 128.41, 128.38, 127.80, 127.65, 127.61, 127.28, 127.20, 127.18, 127.08, 126.92, 126.65, 126.36, 126.30, 126.26, 122.97, 122.94, 121.73, 121.70, 49.34, 22.53, 22.16;如图2所示。
[0031]
实施例2:化合物1b的制备,包括:
具体合成路线如下:。
[0032]
将0.19 mmol三氯化磷溶解于超干dcm(0.7 ml)中。将反应冷却至0
°
c,并向其中逐滴滴加0.93 mmol et3n。10 min后,将反应体系升至室温,并添加0.19 mmol二异丙胺,然后搅拌5 h。在室温下向体系中加入0.19 mmolvapol搅拌反应12 h后,室温条件下加入0.57 mmol硫粉,搅拌反应12小时。反应完成后用硅藻土过滤,并在真空中浓缩、旋干。粗产物通过硅胶(1:100 etoac:石油醚)纯化,得到白色固体化合物1b,产率为47%。
[0033] 1
h nmr (400 mhz, cdcl3) δ 9.85 (dd, j = 9.1, 5.0 hz, 2h), 8.19-7.92 (m, 2h), 7.82 (dd, j = 8.8, 6.7 hz, 2h), 7.79-7.62 (m, 6h), 7.57 (d, j = 1.4 hz, 1h), 7.50 (d, j = 1.3 hz, 1h), 7.09 (dt, j = 15.3, 7.4 hz, 2h), 6.93 (dt, j = 19.0, 7.6 hz, 4h), 6.63-6.44 (m, 4h), 3.45 (dq, j = 21.8, 6.8 hz, 2h), 1.13-0.74 (m, 12h);如图3所示;
13
c nmr (101 mhz, cdcl3) δ 149.76, 149.61, 147.53, 147.44, 141.74, 141.72, 141.49, 141.47, 139.77, 134.70, 134.36, 134.34, 133.45, 133.14, 129.75, 129.58, 129.51, 129.45, 129.26, 129.16, 129.10, 128.86, 128.84, 128.72, 128.16, 128.14, 127.80, 127.64, 127.24, 127.11, 127.02, 126.99, 126.93, 126.90, 126.72, 126.66, 126.47, 125.90, 125.87, 122.98, 122.95, 121.72, 121.69, 49.08, 22.53, 22.30;如图4所示。
[0034]
实施例3:化合物2a的制备,包括:具体合成路线如下:。
[0035]
将0.1 mmol三氯化磷溶解于超干dcm(0.4 ml)中。将反应冷却至0
°
c,并向其中逐滴滴加0.47 mmol et3n。10 min后,将反应体系升至室温,并添加0.1 mmol二甲胺,然后搅拌5 h。在室温下向体系中加入0.1 mmolvapol搅拌反应12 h后,室温条件下加入0.3 mmol硒粉,搅拌反应12小时。反应完成后用硅藻土过滤,并在真空中浓缩、旋干。粗产物通过硅胶
(1:100 etoac:石油醚)纯化,得到白色固体化合物2a,产率为77%。
[0036]1(400 mhz, cdcl3) δ 9.84-9.75 (m, 1h), 9.75-9.61 (m, 1h), 7.97 (ddd, j = 13.1, 7.9, 1.6 hz, 2h), 7.90-7.79 (m, 3h), 7.77-7.64 (m, 5h), 7.59 (dd, j = 5.7, 1.5 hz, 2h), 7.18-7.02 (m, 2h), 7.01-6.87 (m, 4h), 6.54 (ddd, j = 8.3, 6.8, 1.4 hz, 4h), 2.37 (d, j = 11.4 hz, 6h);如图5所示;
13
c nmr (101 mhz, cdcl3) δ 148.66, 148.51, 147.72, 147.63, 141.61, 141.59, 141.30, 141.28, 139.66, 139.48, 134.81, 134.79, 134.53, 134.51, 133.45, 133.31, 129.68, 129.40, 129.37, 129.35, 129.26, 129.21, 128.96, 128.82, 128.68, 128.19, 128.17, 127.84, 127.81, 127.78, 127.76, 127.71, 127.38, 127.16, 127.03, 126.99, 126.94, 126.76, 126.72, 126.65, 126.63, 122.74, 122.70, 121.94, 121.91, 38.82, 38.78;如图6所示。
[0037]
实施例4:化合物2b的制备,包括:具体合成路线如下:。
[0038]
将0.09 mmol三氯化磷溶解于超干dcm(0.4 ml)中。将体系溶液冷却至0
°
c,并向其中逐滴加入0.47 mmol et3n。10 min后,将反应混合物缓慢升至室温,并向其中加入0.09 mmol二甲胺,然后搅拌反应5 h。在室温下再加入0.09 mmol vapol搅拌反应12 h后,向体系中加入0.28 mmol硫粉,搅拌反应12 h。反应完成后用硅藻土过滤,并在真空中浓缩旋干。粗产物在硅胶(1:60 etoac:石油醚)上纯化,得到白色固体的化合物2b,产率为78%。
[0039]
1 (400 mhz, cdcl3) δ 9.77 (d, j = 8.6 hz, 1h), 9.69 (d, j = 8.6 hz, 1h), 7.97 (ddd, j = 13.4, 7.9, 1.6 hz, 2h), 7.83 (m, 3h), 7.77-7.64 (m, 5h), 7.57 (d, j = 4.8 hz, 2h), 7.10 (m, 2h), 6.93 (q, j = 7.9 hz, 4h), 6.54 (t, j = 7.1 hz, 4h), 2.35 (d, j = 11.0 hz, 6h);如图7所示;
13
c nmr (101 mhz, cdcl3) δ 148.79, 148.66, 147.73, 147.65, 141.58, 141.26, 139.72, 139.56, 134.80, 134.50, 133.50, 133.34, 129.68, 129.43, 129.38, 129.32, 129.22, 128.97, 128.94, 128.87, 128.83, 128.15, 128.11, 127.84, 127.77, 127.71, 127.68, 127.35, 127.13, 127.05, 126.96, 126.92, 126.83, 126.72, 126.50, 122.76, 122.05, 122.02, 89.20, 38.59, 38.55;如图8所示。
[0040]
实施例5:化合物3a的制备,包括:具体合成路线如下:
hz, 2h), 7.88-7.61 (m, 8h), 7.60-7.47 (m, 2h), 7.10 (dt, j = 11.9, 7.4 hz, 2h), 6.94 (dt, j = 14.9, 7.6 hz, 4h), 6.53 (d, j = 7.6 hz, 4h), 2.72
ꢀ–ꢀ
2.43 (m, 4h), 1.71 (dt, j = 13.6, 6.8 hz, 2h), 0.72 (d, j = 6.6 hz, 6h), 0.55 (d, j = 6.5 hz, 6h);如图11所示;
13
c nmr (101 mhz, cdcl3) δ 149.89, 149.75, 147.73, 147.64, 141.75, 141.31, 141.29, 139.74, 139.68, 134.74, 134.46, 133.42, 133.25, 129.74, 129.51, 129.42, 129.20, 129.16, 129.15, 129.10, 128.80, 128.77, 128.60, 128.10, 128.07, 127.84, 127.67, 127.62, 127.30, 127.08, 127.05, 127.02, 126.89, 126.71, 126.64, 126.59, 126.05, 126.02, 122.74, 122.71, 122.04, 122.00, 57.54, 57.52, 28.15, 28.12, 20.51, 20.39;如图12所示。
[0046]
实施例7:化合物4a的制备,包括:具体合成路线如下:。
[0047]
将0.09 mmol三氯化磷溶解于超干dcm(0.4 ml)中。将体系溶液冷却至0
°
c,并向其中逐滴加入0.47 mmol et3n。10 min后,将反应混合物缓慢升至室温,并向其中加入0.09 mmol二乙基胺,然后搅拌反应5 h。在室温下再加入0.09 mmol vapol搅拌反应12 h后,向体系中加入0.28 mmol硒粉,搅拌反应12 h。反应完成后用硅藻土过滤,并在真空中浓缩旋干。粗产物在硅胶(1:50 etoac:石油醚)上纯化,得到白色固体的化合物4a,产率为88%。
[0048]
1 (400 mhz, cdcl3) δ 9.76 (ddd, j = 26.4, 8.4, 1.2 hz, 2h), 7.97 (dd, j = 8.0, 1.6 hz, 2h), 7.82 (m, 3h), 7.76-7.65 (m, 5h), 7.59 (dd, j = 11.5, 1.4 hz, 2h), 7.17-7.03 (m, 2h), 7.01-6.86 (m, 4h), 6.56 (td, j = 8.3, 1.3 hz, 4h), 2.95 (dqd, j = 14.2, 7.1, 5.0 hz, 4h), 0.74 (t, j = 7.1 hz, 6h);如图13所示;
13
c nmr (101 mhz, cdcl3) δ 149.00, 148.85, 147.70, 147.61, 141.61, 141.59, 141.35, 141.33, 139.65, 139.56, 134.73, 134.72, 134.42, 134.40, 133.44, 133.19, 129.68, 129.44, 129.42, 129.28, 129.21, 129.10, 129.00, 128.82, 128.79, 128.56, 128.08, 128.06, 127.81, 127.67, 127.50, 127.48, 127.29, 127.08, 126.96, 126.90, 126.78, 126.70, 126.66, 126.48, 126.45, 122.73, 122.70, 121.96, 121.93, 42.44, 42.40, 13.91, 13.89;如图14所示。
[0049]
实施例8:化合物4b的制备,包括:
具体合成路线如下:。
[0050]
将0.09 mmol三氯化磷溶解于超干dcm(0.4 ml)中。将体系溶液冷却至0
°
c,并向其中逐滴加入0.47 mmol et3n。10 min后,将反应混合物缓慢升至室温,并向其中加入0.09 mmol二乙基胺,然后搅拌反应5 h。在室温下再加入0.09 mmol vapol搅拌反应12 h后,向体系中加入0.28 mmol硫粉,搅拌反应12 h。反应完成后用硅藻土过滤,并在真空中浓缩旋干。粗产物在硅胶(1:60 etoac:石油醚)上纯化,得到白色固体的化合物4b,产率为85%。
[0051]1(400 mhz, cdcl3) δ 9.78 (ddd, j = 24.5, 8.7, 1.2 hz, 2h), 7.98 (dd, j = 7.4, 1.7 hz, 2h), 7.89-7.65 (m, 8h), 7.59 (dd, j = 9.8, 1.4 hz, 2h), 7.18-7.05 (m, 2h), 6.96 (dt, j = 11.4, 7.8 hz, 4h), 6.57 (td, j = 8.1, 1.4 hz, 4h), 2.93 (dq, j = 13.9, 7.0 hz, 4h), 0.76 (t, j = 7.1 hz, 6h);如图15所示;
13
c nmr (101 mhz, cdcl3) δ 149.16, 149.02, 147.72, 147.63, 141.58, 141.56, 141.32, 141.30, 139.69, 139.63, 134.72, 134.71, 134.40, 134.38, 133.48, 133.20, 129.67, 129.46, 129.42, 129.25, 129.17, 129.13, 128.86, 128.79, 128.53, 127.98, 127.96, 127.80, 127.68, 127.61, 127.42, 127.39, 127.25, 127.05, 126.98, 126.88, 126.81, 126.71, 126.69, 126.53, 126.51, 126.33, 126.31, 122.73, 122.70, 122.00, 121.97, 42.13, 42.09, 14.00, 13.97;如图16所示。
[0052]
实施例9:硫芳基参与的六元环内酯产物的制备方法,包括:具体合成路线如下:。
[0053]
无水无氧条件下,化合物5a(cas号: 119259-96-0)和化合物5b(cas号: 2762298-39-3),催化剂(化合物4a)和甲基磺酸加入干燥过的反应管中,于0℃低温下,向反应管中加入1.0 ml超干二氯甲烷,氩气氛围中进行反应24小时后,对反应体系进行柱层析纯化,制得
硫芳基参与的六元环内酯产物5c。
[0054]
1 (400 mhz, cdcl3) δ 7.27 (m, 2h), 7.14 (dd, j = 8.6, 4.2 hz, 2h), 6.86 (m, 2h), 6.65 (d, j = 4.2 hz, 2h), 5.17 (dd, j = 7.1, 4.2 hz, 1h), 3.79 (dd, j = 13.7, 4.2 hz, 6h), 3.32 (tt, j = 7.2, 4.2 hz, 1h), 2.85 (dtd, j = 14.8, 7.0, 3.6 hz, 1h), 2.66
ꢀ–ꢀ
2.49 (m, 1h), 2.42 (d, j = 4.2 hz, 6h), 2.14-1.77 (m, 2h);如图17所示;
13
c nmr (101 mhz, cdcl3) δ 170.49, 159.96, 159.86, 145.26, 130.38, 127.86, 121.65, 114.06, 114.03, 83.97, 55.45, 55.28, 46.56, 28.47, 24.20, 22.50;如图18所示。
[0055]
hplc分离对映体:chiralcel
®
ic柱,30℃,正己烷:i-proh = 65:35,1 ml/min,次要保留时间27.57 min,主要保留时间25.27 min,er =60:40;如图21、22所示。
[0056]
实施例10:轴手性含硫双苯基衍生物的制备方法,包括:具体合成路线如下:。
[0057]
无水无氧条件下,化合物6a(cas号: 2411582-25-5)和化合物6b(cas号: 2376073-20-8)催化剂(化合物1a)和对氯苯磺酸加入干燥过的反应管中,于-40℃低温下,向反应管中加入0.5 ml超干二氯甲烷,氩气氛围中进行反应24小时后,将体系升至-20℃,继续反应4小时后,对反应体系进行柱层析纯化,制得轴手性含硫双苯基衍生物6c。
[0058]
1 (400 mhz, cdcl3) δ 7.61-7.52 (m, 1h), 7.44-7.31 (m, 3h), 7.19-7.06 (m, 3h), 6.98 (d, j = 8.5 hz, 1h), 6.48 (d, j = 8.7 hz, 1h), 6.21 (s, 1h), 4.85 (s, 1h), 2.47 (s, 6h);如图19所示;
13
c nmr (101 mhz, cdcl3) δ 153.63, 153.04, 142.38, 135.57, 132.82, 132.76, 132.72, 131.16, 130.43, 130.34, 128.86, 128.75, 127.60, 113.33, 112.10, 108.96, 22.21;如图20所示。
[0059]
hplc分离对映体:chiralcel
®
ic柱,30℃,正己烷:i-proh = 95:5,1 ml/min,次要保留时间6.59 min min,主要保留时间7.16 min,er =48:52;如图23、24所示。
[0060]
对比例1:将如下结构式的硒催化剂作为对比例1:
;其制备方法参考cn114870892a中的实施例1(1c)。
[0061]
【催化性能测试与表征】1. 催化硫芳基参与的六元环内酯产物合成的测试结果测试试样:实施例1-8中基于vapol合成硒硫类化合物以及对比例1中的硒化合物催化剂;其应用实施方案均与实施例9相同,得到的六元环内酯产物的产率(yield)与对映选择性(ee值)如表1所示。
[0062]
表1 六元环内酯产物的产率与ee值由表1可以看出,化合物1a、化合物1b、化合物2a、化合物2b、化合物3a、化合物3b、化合物4a、化合物4b催化硫芳基参与的六元环内酯产物合成的产率≥94%,ee值≥20%,相对于对比例1,本发明制得的基于vapol合成的硒硫类化合物对硫芳基参与的六元环内酯产物具有优良的催化合成效果,且能够得到对映选择性较高的硫芳基参与的六元环内酯产物。
[0063]
2. 催化轴手性含硫双苯基衍生物合成的测试结果测试试样:实施例1-8中基于vapol合成硒硫类化合物以及对比例1中的硒化合物催化剂;其应用实施方案均与实施例10相同,得到的六元环内酯产物的产率(yield)与对映选择性(ee值)如表2所示。
[0064]
表2 轴手性含硫双苯基衍生物的产率与ee值由表2可以看出,化合物1a、化合物1b、化合物2a、化合物2b、化合物3a、化合物3b、化合物4a、化合物4b催化轴手性含硫双苯基衍生物的产率≥75%,ee值≥4%,相对于对比例
1,本发明制得的基于vapol合成的硒硫类化合物对轴手性含硫双苯基衍生物具有优良的催化合成效果,能够得到一定对映选择性的轴手性含硫双苯基衍生物。
[0065]
上述的对实施例的描述是为便于该技术领域的普通技术人员能理解和使用发明。熟悉本领域技术的人员显然可以容易地对这些实施例做出各种修改,并把在此说明的一般原理应用到其他实施例中而不必经过创造性的劳动。因此,本发明不限于上述实施例,本领域技术人员根据本发明的揭示,不脱离本发明范畴所做出的改进和修改都应该在本发明的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1