一种导电MOFs均匀包覆的Ti:Fe2O3复合光阳极及其制备方法和应用
一种导电mofs均匀包覆的ti:fe2o3复合光阳极及其制备方法和应用
技术领域
1.本发明涉及纳米材料制备和光电催化技术领域,具体涉及一种导电mofs包覆的ti:fe2o3复合光阳极及其制备方法和在光电催化水氧化中的应用。
背景技术:
2.光电催化分解水制氢技术的应用被认为是解决能源问题的一种有效途径。然而,分解水制氢技术分为两个半反应:水还原制氢,水氧化制氧。其中,水氧化制氧为四电子过程较为缓慢成为水分解的速控步骤。因此,高效稳定的水氧化催化剂的设计与制备是分解水制氢技术的关键。氧化铁(fe2o3)作为一种新型的可见光响应光催化剂,具有独特的电子结构、较窄的禁带宽度和合适的能带位置,并具有较高的热稳定性和化学稳定性,被广泛的应用于光电催化分解水的研究。但其光生电子和空穴的快速复合较为严重,并且对水的氧化能力不足制约了fe2o3的实际应用。研究者采用了大量的方法对fe2o3进行了改性研究,其中包括元素掺杂(ti
1.,zr
2.,sn
3.),氧空位的引入
4.,助催化剂表面修饰
5.等。元素掺杂能够促进fe2o3的导电性,进而促进光生电子-空穴的分离。另外,助催化剂的表面修饰是促进电子-空穴分离和提高产氧动力学的有效方法。但是文献报道的多数助催化剂以颗粒的形式负载到fe2o3的表面,助催化剂与fe2o3接触面较小,不利于电子-空穴的高效分离。核壳结构的设计能够实现高效的电子传输,提高光阳极的活性和稳定性。但是目前制备核壳结构光阳极的方法较为单一,主要是光沉积法,表面活性剂辅助的溶剂热法。上述两种方法存在较大问题,其中光沉积法只适用于几类核壳结构的制备,普适性较差。另外,表面活性剂作为表面配位基团合成的核壳结构光阳极的稳定性较差。
3.[1]z.-y.wang,h.-m.li,s.-s.yi,m.-z.you,h.-j.jing,x.-z.yue,z.-t.zhang,d.-l.chen,in-situcoatingofmultifunctionalfeco-bimetalorganicframeworknanolayersonhematitephotoanodeforsuperioroxygenevolution,appl.catal.b:environ.,297(2021)120406.
[0004]
[2]s.shen,p.guo,d.a.wheeler,j.jiang,s.a.lindley,c.x.kronawitter,j.z.zhang,l.guo,s.s.mao,physicalandphotoelectrochemicalpropertiesofzr-dopedhematitenanorodarrays,nanoscale,5(2013)9867-9874.
[0005]
[3]a.g.hufnagel,h.hajiyani,s.zhang,t.li,o.kasian,b.gault,b.breitbach,t.bein,d.fattakhova-rohlfing,c.scheu,r.pentcheva,whytin-dopingenhancestheefficiencyofhematitephotoanodesforwatersplitting-thefullpicture,adv.funct.mater.,28(2018)1804472.
[0006]
[4]h.-m.li,z.-y.wang,h.-j.jing,s.-s.yi,s.-x.zhang,x.-z.yue,z.-t.zhang,h.-x.lu,d.-l.chen,synergeticintegrationofpassivationlayerandoxygenvacancyonhematitenanoarraysforboostedphotoelectrochemicalwateroxidation,appl.catal.b:environ.,284(2021)119760.
[0007]
[5]j.-b.pan,s.shen,l.chen,c.t.au,s.f.yin,core
–
shell photoanodes for photoelectrochemical water oxidation,adv.funct.mater.,(2021)2104269.
技术实现要素:
[0008]
为了解决现有技术中存在的问题,本发明的目的是在于提供了一种导电 mofs均匀包覆的ti:fe2o3复合光阳极及其制备方法和应用,通过溶剂热法将导电mofs包覆在经表面阴离子化处理后的ti:fe2o3的表面,导电mofs的表面包覆一方面可以调控ti:fe2o3的表面态,快速转移其与ti:fe2o3界面的空穴,实现高效的电荷分离;另外,导电mofs的水氧化过电势更低,更有利于水氧化过程。
[0009]
为了实现上述技术目的,本发明采用如下技术方案:
[0010]
一种导电mofs包覆的ti:fe2o3复合光阳极,具有“核壳结构”,“核”为表面阴离子化的ti:fe2o3纳米片阵列,“壳”为导电mofs;
[0011]
所述表面阴离子化用的阴离子表面活性剂为含有邻位酚羟基的羧酸或者含有邻位二氨基的羧酸。
[0012]
优选的,所述含有邻位酚羟基的羧酸为单宁酸、没食子酸、咖啡酸、3,4-二羟基苯甲酸或2,3,4-三羟基苯甲酸,所述含有邻位二氨基的羧酸为3,4-二氨基苯甲酸。
[0013]
优选的,所述导电mofs的厚度为1-3nm。
[0014]
优选的,所述导电mofs为单金属-有机骨架或双金属-有机骨架,单金属为钴、铁或镍;双金属为镍铁、镍钴或钴铁。
[0015]
更优选的,所述导电mofs为镍铁双金属-有机骨架,镍铁的摩尔比为2~4:1;或为钴铁双金属-有机骨架,钴铁的摩尔比为2~4:1。
[0016]
需要说明的是,本发明中的ti:fe2o3纳米片阵列和导电mofs均采用现有常规技术制得,并无特殊要求,在此不再赘述。
[0017]
本发明还提供了上述导电mofs包覆的ti:fe2o3复合光阳极的制备方法,先将ti:fe2o3纳米片阵列置于阴离子表面活性剂的醇溶液中进行表面阴离子化处理后,再通过溶剂热法将导电mofs包覆到ti:fe2o3表面即可。
[0018]
本发明中的ti:fe2o3纳米阵列可参照现有技术制得,例如水热法-热处理方式,具体制备过程为:将2.43g六水合三氯化铁(fecl3·
6h2o)和0.811g尿素溶于90ml超纯水中(控制水温在5℃以下)。搅拌15分钟后,向上述溶液中缓慢滴加450μl 0.1m四氯化钛(ticl4)乙醇溶液。搅拌30分钟后,将混合液和处理过的导电玻璃(fto,氟掺杂氧化锡作为导电层,导电面向下)转移至晶化釜,110℃下晶化4h。将晶化釜冷却至室温后,用超纯水冲洗电极三次。然后,依次将电极80℃干燥12h,550℃退火2h,600℃退火20min(升温速率为 5℃/min),得到电极为ti:fe2o3纳米阵列。
[0019]
优选的,所述阴离子表面活性剂的醇溶液的浓度为2~10mg/ml。
[0020]
优选的,所述导电mofs为单金属-有机骨架或双金属-有机骨架,单金属为钴、铁或镍;双金属为镍铁、镍钴或钴铁。
[0021]
更优选的,所述mofs为镍铁双金属-有机骨架,镍铁的摩尔比为2~4:1,进一步优选为3:1;或为钴铁双金属-有机骨架,钴铁的摩尔比为2~4:1,进一步优选为3:1。发明人发现,与单金属导电mof相比,双金属导电mofs具有更好的导电性和更丰富的活性位点。
[0022]
本发明中通过溶剂热法将导电mofs包覆到ti:fe2o3表面可参照现有技术制得,先制得导电mofs的前驱体溶液,再将ti:fe2o3置于前驱体溶液中,经溶剂热处理后,再经醇洗、干燥后即得导电mofs包覆的ti:fe2o3复合光阳极。
[0023]
本发明还提供了上述导电mofs包覆的ti:fe2o3复合光阳极的应用,将其用于光电催化水氧化。
[0024]
本发明采用含有邻位酚羟基的羧酸(例如单宁酸、咖啡酸、3,4-二羟基苯甲酸、2,3,4-三羟基苯甲酸、没食子酸)或者含有邻位二氨基的羧酸(例如3,4-二氨基苯甲酸)为表面配位基团,构筑了ti:fe2o3@导电mofs。基于邻羟基或者邻氨基与ti:fe2o3的配位作用,单宁酸、咖啡酸、3,4-二羟基苯甲酸、2,3,4-三羟基苯甲酸、没食子酸或者3,4-二氨基苯甲酸能够牢牢地锚定在ti:fe2o3的表面。同时ni
2+
、fe
2+
或者co
2+
与单宁酸、咖啡酸、3,4-二羟基苯甲酸、2,3,4-三羟基苯甲酸和没食子酸或者3,4-二氨基苯甲酸上的羧基配位,在溶剂热条件下,可以形成均匀的ti:fe2o3@导电mofs核壳结构光阳极。导电mofs的表面包覆一方面可以调控ti:fe2o3的表面态,实现高效的电荷分离;另外,导电mofs的水氧化过电势更小,fe,ni或者co作为活性位点能够有效地促进水的氧化,提高水分解效率。
[0025]
与现有技术相比,本发明的优点:
[0026]
本发明首先以简单的水热法-热处理法制备了ti:fe2o3纳米棒阵列,有效防止了ti:fe2o3棒堆积。ti掺杂能够有效提高fe2o3导电性。然后采用含有邻位酚羟基的羧酸(例如单宁酸、咖啡酸、3,4-二羟基苯甲酸、2,3,4-三羟基苯甲酸、没食子酸)或者含有邻位二氨基的羧酸(例如3,4-二氨基苯甲酸)作为表面修饰剂,基于邻位酚羟基或者邻位氨基与ti:fe2o3的配位作用以及羧基与金属离子的配位作用,采用溶剂热法在ti:fe2o3表面均匀包覆导电mofs。导电mofs的表面包覆一方面通过调控ti:fe2o3的表面态增强ti:fe2o3与导电mofs界面的导电性,同时金属元素(co、ni、fe元素)作为活性位点能够有效促进水的氧化,增强光电催化水分解活性。
附图说明
[0027]
图1为实施例8(a)、实施例1(b)、实施例9(c)和实施例10(d)制得的样品 b1,tfdm-0,b2,b3的tem图;
[0028]
图2为实施例1制得的ti:fe2o3、tfdm-0和实施例9制得的b2样品的xrd 图;
[0029]
图3为实施例1制得的ti:fe2o3、tfdm-0和实施例9制得的b2样品的光电流-电压(j-v)曲线。
[0030]
图4为实施例1制得的ti:fe2o3、tfdm-0和实施例9制得的b2样品的的交流阻抗(eis)谱。
[0031]
如图1所示,基于咖啡酸的表面配位作用,导电cofe-mofs均匀地包覆在ti:fe2o3的表面形成核壳结构。
[0032]
如图2所示,水热法制备的ti:fe2o3为六方晶型。与ti:fe2o3相比,晶化6h 得到的ti:fe2o3@cofe-mofs中并没有观察到cofe-mofs的特征衍射峰,说明表面包覆的cofe-mofs很薄且均匀分布。而晶化12h得到的 ti:fe2o3@cofe-mofs(b2)中则明显可以观察到cofe-mofs的特征衍射峰。
[0033]
如图3所示,与ti:fe2o3相比,表面包覆cofe-mofs以后, ti:fe2o3@cofe-mofs的活
性得到明显提高。其中,晶化6h得到的 ti:fe2o3@cofe-mofs(tfdm-0)具有最佳的光电催化活性,1.23v vs rhe电压下电流密度达到3.3macm-2
。
[0034]
如图4所示,与ti:fe2o3相比,ti:fe2o3@cofe-mofs的阻抗明显变小,说明基于咖啡酸的配作作用,ti:fe2o3与cofe-mofs界面形成的紧密接触能够有效促进电子-空穴的转移。
具体实施方式
[0035]
下面结合实施例对本发明做进一步详细说明,但本发明的保护范围并不局限于这些实施例。
[0036]
本发明通过光电催化水氧化来评价ti:fe2o3@导电mofs的活性,所用的电解质溶液为ph=13.6的1m氢氧化钾(koh)溶液;光源采用300w氙灯配有 am 1.5滤光片,通过控制光源与光阳极的距离,使光强为100mw/cm2;外加电源通过辰华660e电化学工作站提供。反应产生的氢气和氧气采用气相色谱定量。
[0037]
本发明以改变cofe-mofs中金属元素比例,表面改性剂浓度及其种类和晶化时间在ti:fe2o3表面的包覆导电mofs,制备复合光阳极。
[0038]
实施例1
[0039]
ti:fe2o3纳米棒阵列的制备:将2.43g六水合三氯化铁(fecl3·
6h2o)和0.811 g尿素溶于90ml超纯水中(控制水温在5℃以下,防止后续加入的四氯化钛快速水解),搅拌15分钟。向上述溶液中缓慢滴加450μl 0.1m四氯化钛(ticl4) 乙醇溶液,搅拌30分钟。将混合液和处理过的导电玻璃(fto,氟掺杂氧化锡作为导电层,导电面向下)转移至晶化釜,110℃下晶化4h。将晶化釜冷却至室温后,用超纯水冲洗电极三次。然后,依次将电极80℃干燥12h,550℃退火2 h,600℃退火20min(升温速率为5℃/min),得到电极为ti:fe2o3纳米阵列。
[0040]
ti:fe2o3表面处理:将ti:fe2o3纳米棒阵列置于5mg/ml咖啡酸乙醇溶液中,在50℃下浸泡3h,然后用无水乙醇冲洗一次,备用。
[0041]
核壳结构ti:fe2o3@导电mofs的制备:将36mg四水合醋酸钴 (co(oac)2·
4h2o),13.4mg七水合硫酸亚铁(feso4·
7h2o)溶解于5ml n,n-二甲基甲酰胺(dmf)中,搅拌30min,命名为a溶液。将36mg 2,5-二羟基-1,4-对苯醌和300mg四丁基溴化铵溶于12mldmf,3ml乙醇和3ml水中,在氮气七分钟搅拌15min,命名为b溶液。然后将上述a溶液逐滴滴加到b溶液中,混合均匀。将混合液和阴离子化的ti:fe2o3纳米棒阵列转移到玻璃瓶中,100℃晶化6h。之后,将晶化釜自然冷却至室温,分别用dmf、乙醇、去离子水冲洗三次后,浸没在丙酮中24h,除去导电mofs孔道中的dmf。进一步置于烘箱中80℃干燥12h,所得样品记为tfdm-0。
[0042]
以光电催化水氧化为模型反映考察所制备的光阳极的催化活性:
[0043]
所用的电解质溶液为ph=13.6的1m koh溶液。光源采用300w氙灯配有 am 1.5滤光片,通过控制光源与光阳极的距离,使照射到光阳极的光强为100 mw/cm2,光照时间为4h。外加电源通过辰华660e电化学工作站提供。每光照 30min,通过进样环将产生的部分氢气和氧气打入气相色谱(gc2010,热导检测器,安捷伦公司产)定量。外加偏压为1.23v vs rhe,光照4小时氢气和氧气的产生速率分别为60.1μmol h-1
cm-2
和31.5μmol h-1
cm-2
。
[0044]
实施例2~4
[0045]
对cofe-mofs中co:fe不同摩尔比的ti:fe2o3@导电mofs复合光催化剂,操作步骤同实施例1,只改变溶剂热过程中co(oac)2·
4h2o与feso4·
7h2o加入的量,其余条件均不变,
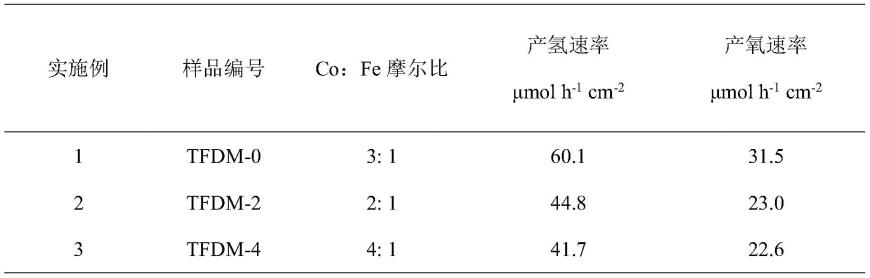
并把样品编号为tfdm-2、tfdm-4,其结果见如表1 所示。
[0046]
表1co:fe不同摩尔比的ti:fe2o3@导电mofs复合光阳极的反应结果
[0047][0048]
由表1可知,在不同co、fe摩尔比下得到不同的水分解速率,其中co、 fe摩尔比为3:1时产氢速率为60.1μmol h-1
cm-2
,产氧速率为31.5μmol h-1
cm-2
,水氧化效果最好。
[0049]
实施例5~7
[0050]
按照效果最优的的实施例1的步骤,其余条件不变(co:fe的摩尔比为3:1),只改变咖啡酸的浓度,分别变为2mg/ml、7mg/ml、10mg/ml,并将其样品编号为a1、a2、a3,其结果见如表2所示。
[0051]
表2不同咖啡酸浓度所得ti:fe2o3@导电mofs复合光阳极的反应结果
[0052][0053]
由表2可得,在不同的咖啡酸浓度修饰下得到催化活性不同的光阳极材料,咖啡酸的浓度为5mg/ml时所制备的核壳结构ti:fe2o3@导电mofs光阳极具有最佳的光电催化水氧化效果。
[0054]
实施例8~10
[0055]
同实施例1,只改变晶化时间为3h,12h,18h,并把其样品编号为b1、 b2、b3,其结果见如表3所示。
[0056]
表3不同晶化时间所得ti:fe2o3@导电mofs复合光阳极的反应结果
[0057][0058]
由表3可得,在不同的咖啡酸浓度修饰下得到催化活性不同的光阳极材料,当晶化时间为6h时所制备的核壳结构ti:fe2o3@导电mofs光阳极具有最佳的光电催化水氧化效果。
[0059]
实施例11~15
[0060]
同实施例1,只改变表面改性剂的种类为单宁酸、没食子酸、3,4-二羟基苯甲酸、2,3,4-三羟基苯甲酸和3,4-二氨基苯甲酸,并把其样品编号为c1、c2、c3、 c4、c5,其结果见如表3所示。
[0061]
表4不同表面改性剂种类的ti:fe2o3@导电mofs复合光阳极的反应结果
[0062][0063]
由表4可得,采用不同的表面改性剂所制备的光阳极具有不同的产氢产氧速率,咖啡酸作为表面改性剂制备的光阳极具有最高的产氢产氧速率,光电催化分解水效果最佳。
[0064]
对比例1
[0065]
以水热合成的ti:fe2o3纳米阵列作为光阳极评价其光电催化水氧化活性,所得样品为tf。具体操作和活性评价方法与实施例1一致,外加偏压为1.23v vs rhe,光照4小时氢气和氧气的产生速率分别为19.5μmol h-1
cm-2
和10.2μmol h-1
cm-2
。
[0066]
对比例2
[0067]
以2,5-二羟基-1,4-对苯醌为配体制备导电cofe-mofs光阳极。具体操作如下:将36mg四水合醋酸钴(co(oac)2·
4h2o),13.4mg七水合硫酸亚铁 (feso4·
7h2o)溶解于5ml n,n-二甲基甲酰胺(dmf)中,搅拌30min,命名为a 溶液。将36mg 2,5-二羟基-1,4-对苯醌和
300mg四丁基溴化铵溶于12mldmf, 3ml乙醇和3ml水中,在氮气七分钟搅拌15min,命名为b溶液。然后将上述a溶液逐滴滴加到b溶液中,混合均匀。将混合液和阴离子化(阴离子表面活性剂为5mg/ml咖啡酸乙醇溶液)的fto玻璃转移到玻璃瓶中,100℃晶化6h。之后,将晶化釜自然冷却至室温,分别用dmf、乙醇、去离子水冲洗三次后,浸没在丙酮中24h,除去导电mofs孔道中的dmf。进一步置于烘箱中 80℃干燥12h,所得样品记为dcfm。活性评价方法与实施例1一致,外加偏压为1.23v vs rhe,光照4小时氢气和氧气的产生速率分别为~0.1μmol h-1
cm-2
和~0.1μmol h-1
cm-2
。
[0068]
对比例3
[0069]
ti:fe2o3纳米棒阵列的制备同实施例1。
[0070]
ti:fe2o3表面处理:将ti:fe2o3纳米棒阵列置于5mg/ml聚乙烯吡咯烷酮 (pvp k30)乙醇溶液中,在50℃下浸泡3h,然后用无水乙醇冲洗一次,备用。
[0071]
2,5-二羟基-1,4-对苯醌为配体制备复合光阳极:将36mg四水合醋酸钴 (co(oac)2·
4h2o),13.4mg七水合硫酸亚铁(feso4·
7h2o)溶解于5ml n,n-二甲基甲酰胺(dmf)中,搅拌30min,命名为a溶液。将36mg 2,5-二羟基-1,4-对苯醌和300mg四丁基溴化铵溶于12mldmf,3ml乙醇和3ml水中,在氮气七分钟搅拌15min,命名为b溶液。然后将上述a溶液逐滴滴加到b溶液中,混合均匀。将混合液和pvp k30阴离子化的ti:fe2o3纳米棒阵列转移到玻璃瓶中,100℃晶化6h。之后,将晶化釜自然冷却至室温,分别用dmf、乙醇、去离子水冲洗三次后,浸没在丙酮中24h,除去导电mofs孔道中的dmf。进一步置于烘箱中80℃干燥12h,所得样品记为tfdm-p。活性评价方法与实施例1 一致,外加偏压为1.23vvs rhe,光照4小时氢气和氧气的产生速率分别为28.1 μmol h-1
cm-2
和14.9μmol h-1
cm-2
。
[0072]
表5实施例1和对比例1-3的反应结果
[0073][0074]
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1