一种苯并磺内酰胺类化合物的电化学制备方法
1.本发明属于苯并磺内酰胺类化合物的合成技术领域,具体涉及一种以电化学方法,在无催化剂、氧化剂的条件下来实现5
‑
、6
‑
和7
‑
元的苯并磺内酰胺类化合物的制备。
背景技术:
2.磺胺类化合物是广谱的抗菌类化合物。作为环状的磺胺,苯并磺内酰胺类化合物具有广泛的生物活性和药理活性,包括抗菌、治疗糖尿病、抗肿瘤、抗抑郁和抗病毒等作用。此外,苯并磺内酰胺类化合物还可以作为手性辅基被用于不对称催化中。传统的制备苯并磺内酰胺类化合物的方法主要有:偶极环加成、亲核加成、过渡金属催化的c
‑
h键活化、corey
–
bakshi
–
shibata还原、aza
‑
darzens缩合等等。虽然这些策略提供了有效的途径获得苯并磺内酰胺,但或多或少存在一些局限性,例如:大多数反应涉及使用有害和昂贵的过渡金属催化剂、化学计量氧化剂、苛刻的反应条件等问题。因此,发展一种温和、绿色高效的方法来构建苯并磺内酰胺类化合物具有极大的研究和实用价值(molecules 2020,25,4367)。
3.近年来,由于电化学合成具有环境友好、反应性能优越等优点,引起了广大研究工作者的广泛关注。与传统的合成方法相比,电化学合成避免了危险和有毒的氧化还原试剂的使用。电化学法可以通过阳极氧化及阴极还原,来生成高活性中间体(自由基、自由基离子、阳离子、阴离子等等),因此被认为是一个绿色的和强大的工具来实现碳
‑
碳、碳
‑
杂原子之间共价键的构筑(chem.rev.2008,108,2265.)。在这种策略下,很多具有生物活性的含氮杂环,如吡咯烷酮、吲哚、内酰胺等,可以通过阳极氧化所形成的氮中心自由基及阳离子自由基来实现c
‑
n键的构筑(j.am.chem.soc.2000,122,5636
–
5637)。然而,以电化学方法来制备磺内酰胺还鲜有报道。近来,仅有雷爱文教授小组报道了co催化下通过电化学作用实现磺胺类化合物与炔烃的[4+2]环加成,进而得到苯并磺内酰胺类化合物(green chem.,2020,22,1548
–
1552)。
技术实现要素:
[0004]
本发明的目的是提供一种无金属催化剂、无氧化剂条件下,通过电化学方法来构筑苯并磺内酰胺类化合物的方法。该方法反应条件温和、反应高效、仅通过调整芳基磺内酰胺邻位的取代基,即可在在电化学条件下,实现5
‑
、6
‑
和7
‑
元的苯并磺胺类化合物的制备。
[0005]
针对上述目的,本发明所采用的技术方案是:将式i或ii或iii所示的芳基磺胺类反应底物加入以四正丁基乙酸铵为电解质的电解液中,以碳电极作为阳极、铂片作为阴极,在恒定的电流作用下反应1~2小时,得到式i’或ii’或iii’所示的苯并磺酰胺类化合物;
[0006][0007]
式中r1代表c
1~4
烷基,r2、r3各自独立代表h、c
1~4
烷基、卤素中任意一种或两种,r4代表c
1~4
烷基、卤素、cf3中任意一种或两种,且r4所在苯环的邻位至少有一个为h且另一个邻位不为c
1~4
烷基;r5代表h或c
1~4
烷基。
[0008]
上述制备方法中,优选所述芳基磺胺类反应底物与四正丁基乙酸铵的摩尔比为1:1~3,所述电解液中加入的芳基磺胺类反应底物的浓度为0.01~0.1mol/l,所述电解液是以六氟异丙醇与甲醇或者乙腈体积比为1:2的混合液为溶剂。
[0009]
上述制备方法中,所述的碳电极为碳棒、碳片、碳布、网状玻璃态碳中任意一种。
[0010]
上述制备方法中,优选反应电流为2~20ma。
[0011]
本发明的有益效果如下:
[0012]
本发明将芳基磺胺类反应底物及电解质溶于相应的溶剂中,以碳电极作为阳极,以铂片作为阴极,在恒定的反应电流作用下,通过阳极氧化诱导反应底物产生磺酰氨基自由基,并进一步发生分子内环化,在无金属催化剂和氧化剂的情况下,通过电化学方法实现了苯并磺酰胺类化合物的制备。本发明反应条件温和,反应高效、便捷,底物适用范围广,产物收率高,仅通过调整芳基磺内酰胺邻位的取代基,即可在电化学条件下,实现5
‑
、6
‑
和7
‑
元的苯并磺胺类化合物的制备,具有很好的应用前景。
附图说明
[0013]
图1是实施例1中终产物i
’‑
1的核磁氢谱图。
[0014]
图2是实施例6中终产物ii
’‑
1的核磁氢谱图。
[0015]
图3是实施例11中终产物iii
’‑
1的核磁氢谱图。
具体实施方式
[0016]
下面结合实施例对本发明进一步详细说明,但本发明的保护范围不仅限于这些实
施例。
[0017]
实施例1
[0018]
将60.7mg(0.2mmol)芳基磺胺类反应底物i
‑
1、60.3mg(0.2mmol)四正丁基乙酸铵(n
‑
bu4noac)溶于6.0ml乙腈与六氟异丙醇体积比为2:1的混合溶液中,以6mm碳棒作为阳极,以1cm
×
1cm铂片作为阴极,在15ma电流作用下反应1.5小时(薄层色谱监测反应完全)后,将反应液浓缩,硅胶柱色谱分离(乙酸乙酯/石油醚=1~10,v/v)得到42.2mg产物i
’‑
1,收率为70%;其反应方程式如下:
[0019][0020]
所得产物的结构表征数据为:1h nmr(400mhz,acetone
‑
d6)δ7.76
–
7.71(m,1h),7.63
–
7.60(m,2h),7.58
–
7.52(m,2h),7.40
–
7.35(m,2h),7.31
–
7.26(m,1h),7.24
–
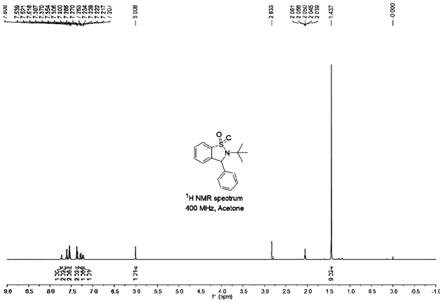
7.21(m,1h),6.01(s,1h),1.44(s,9h),见图1;
13
c nmr(100mhz,cdcl3)δ142.8,137.9,134.4,132.7,129.0,128.9,128.0,126.4,124.8,120.7,63.5,58.6,29.0.
[0021]
实施例2
[0022]
将57.9mg(0.2mmol)芳基磺胺类反应底物i
‑
2、60.3mg(0.2mmol)n
‑
bu4noac溶于6.0ml乙腈与六氟异丙醇体积比为2:1的混合溶液中,以1cm
×
1cm碳布作为阳极,以1cm
×
1cm铂片作为阴极,在15ma电流作用下反应1.5小时(薄层色谱监测反应完全)后,将反应液浓缩,硅胶柱色谱分离(乙酸乙酯/石油醚=1~10,v/v)得到41.4mg产物i
’‑
2,收率为72%;其反应方程式如下:
[0023][0024]
所得产物的结构表征数据为:1h nmr(400mhz,cdcl3)δ7.82
–
7.78(m,1h),7.51
–
7.44(m,2h),7.40
–
7.31(m,5h),7.04
–
7.00(m,1h),5.54(s,1h),3.94(hept,j=6.8hz,1h),1.47(d,j=6.8hz,3h),1.13(d,j=6.8hz,3h).
13
c nmr(100mhz,cdcl3)δ139.6,138.4,134.3,132.7,129.1,129.0,128.7,127.7,125.0,120.8,62.4,46.6,21.9,20.3.hrms(esi)m/z理论值c
16
h
17
no2s[m+na]
+
310.0872,实测值:310.0869.
[0025]
实施例3
[0026]
将64.8mg(0.2mmol)芳基磺胺类反应底物i
‑
3、60.3mg(0.2mmol)n
‑
bu4noac溶于6.0ml乙腈与六氟异丙醇体积比为2:1的混合溶液中,以1cm
×
1cm碳片作为阳极,以1cm
×
1cm铂片作为阴极,在15ma电流作用下反应1.5小时(薄层色谱监测反应完全)后,将反应液浓缩,硅胶柱色谱分离(乙酸乙酯/石油醚=1~10,v/v)得到51.5mg产物i
’‑
3,收率为82%;其反应方程式如下:
[0027][0028]
所得产物的结构表征数据为:1h nmr(400mhz,cdcl3)δ7.83
–
7.79(m,1h),7.53
–
7.47(m,2h),7.35(s,4h),7.04
–
6.98(m,1h),5.52(s,1h),3.94(hept,j=6.8hz,1h),1.46(d,j=6.8hz,3h),1.13(d,j=6.8hz,3h).
13
c nmr(100mhz,cdcl3)δ138.3,137.8,134.7,134.2,132.9,129.4,129.3,129.0,124.9,121.0,61.6,46.7,22.0,20.3.
[0029]
实施例4
[0030]
将61.5mg(0.2mmol)芳基磺胺类反应底物i
‑
4、60.3mg(0.2mmol)n
‑
bu4noac溶于6.0ml乙腈与六氟异丙醇体积比为2:1的混合溶液中,以6mm碳棒作为阳极,以1cm
×
1cm铂片作为阴极,在15ma电流作用下反应1.5小时(薄层色谱监测反应完全)后,将反应液浓缩,硅胶柱色谱分离(乙酸乙酯/石油醚=1~10,v/v)得到48.9mg产物i
’‑
4,收率为80%;其反应方程式如下:
[0031][0032]
所得产物的结构表征数据为:1h nmr(400mhz,cdcl3)δ7.83
–
7.78(m,1h),7.54
–
7.47(m,2h),7.41
–
7.35(m,2h),7.09
–
7.04(m,2h),7.03
–
6.98(m,1h),5.54(s,1h),3.94(hept,j=6.8hz,1h),1.46(d,j=6.8hz,3h),1.12(d,j=6.8hz,3h);
13
c nmr(100mhz,cdcl3)δ162.9(j=246.5hz),138.1,135.5(j=3.2hz),134.2,132.8,129.4,129.3(j=4.9hz),124.9,120.9,116.1(j=21.7hz),61.6,46.6,21.9,20.3.
[0033]
实施例5
[0034]
将64.8mg(0.2mmol)芳基磺胺类反应底物i
‑
5、60.3mg(0.2mmol)n
‑
bu4noac溶于6.0ml乙腈与六氟异丙醇体积比为2:1的混合溶液中,以6mm碳棒作为阳极,以1cm
×
1cm铂片作为阴极,在15ma电流作用下反应1.5小时(薄层色谱监测反应完全)后,将反应液浓缩,硅胶柱色谱分离(乙酸乙酯/石油醚=1~10,v/v)得到53.4mg产物i
’‑
5,收率为83%;其反应方程式如下:
[0035]
[0036]
所得产物的结构表征数据为:1h nmr(600mhz,acetone
‑
d6)δ7.85(d,j=8.4hz,1h),7.64(ddd,j=8.4,2.1,0.6hz,1h).7.55
–
7.54(m,2h),7.45
‑
7.43(m,2h),7.40
‑
7.38(m,1h),7.21
–
7.20(m,1h),5.86(s,1h),3.95(hept,j=6.6hz,1h),1.43(d,j=6.6hz,3h),1.13(d,j=6.6hz,3h);
13
c nmr(150mhz,cdcl3)δ140.4,139.2,138.8,132.8,129.8,129.3,129.1,127.6,125.2,122.2,62.0,46.8,21.8,20.3.
[0037]
实施例6
[0038]
将49.5mg(0.2mmol)芳基磺胺类反应底物ii
‑
1、60.3mg(0.2mmol)n
‑
bu4noac溶于6.0ml乙腈与六氟异丙醇体积比为2:1的混合溶液中,以6mm碳棒作为阳极,以1cm
×
1cm铂片作为阴极,在15ma电流作用下反应1.5小时(薄层色谱监测反应完全)后,将反应液浓缩,硅胶柱色谱分离(乙酸乙酯/石油醚=1~10,v/v)得到33.4mg产物ii
’‑
1,收率为68%;其反应方程式如下:
[0039][0040]
所得产物的结构表征数据为:1h nmr(600mhz,cdcl3)δ8.02
‑
8.01(m,2h),7.97(d,j=7.8hz,1h),7.71(td,j=7.2,1.2hz,1h),7.57(td,j=7.8,1.2hz,1h),7.51(td,j=7.8,1.2hz,1h),7.34(td,j=7.8,1.2hz,1h),7.32(d,j=8.4hz,1h),3.45(s,3h),见图2;
13
c nmr(100mhz,cdcl3)δ139.6,134.3,132.4,132.4,130.4,128.2,125.5,125.4,124.7,124.0,122.5,119.4,32.8.
[0041]
实施例7
[0042]
将55.1mg(0.2mmol)芳基磺胺类反应底物ii
‑
2、60.3mg(0.2mmol)n
‑
bu4noac溶于6.0ml乙腈与六氟异丙醇体积比为2:1的混合溶液中,以1cm
×
1cm网状玻璃态碳作为阳极,以1cm
×
1cm铂片作为阴极,在15ma电流作用下反应1.5小时(薄层色谱监测反应完全)后,将反应液浓缩,硅胶柱色谱分离(乙酸乙酯/石油醚=1~10,v/v)得到44.3mg产物ii
’‑
2,收率为81%;其反应方程式如下:
[0043][0044]
所得产物的结构表征数据为:1h nmr(600mhz,cdcl3)δ7.96
–
7.95(m,2h),7.91(d,j=7.8hz,1h),7.68(td,j=7.8,1.2hz,1h),7.55(td,j=7.2,1.2hz,1h),7.46
–
7.42(m,3h),4.48
–
4.41(m,1h),1.09(d,j=6.8hz,6h);
13
c nmr(150mhz,cdcl3)δ137.2,136.6,133.2,132.4,129.6,128.7,128.4 127.3,127.2,125.7,125.5,123.0,54.9,21.6.
[0045]
实施例8
[0046]
将62.0mg(0.2mmol)芳基磺胺类反应底物ii
‑
3、60.3mg(0.2mmol)n
‑
bu4noac溶于6.0ml乙腈与六氟异丙醇体积比为2:1的混合溶液中,以6mm碳棒作为阳极,以1cm
×
1cm铂片
作为阴极,在15ma电流作用下反应1.5小时(薄层色谱监测反应完全)后,将反应液浓缩,硅胶柱色谱分离(乙酸乙酯/石油醚=1~10,v/v)得到39.4mg产物ii
’‑
3,收率为72%;其反应方程式如下:
[0047][0048]
所得产物的结构表征数据为:1h nmr(400mhz,cdcl3)δ7.95(d,j=7.6hz,1h),7.88
‑
7.84(m,2h),7.68(t,j=7.6hz,1h),7.56(t,j=7.6hz,1h),7.45(s,1h),7.40
‑
7.38(m,1h),4.43((hept,j=6.8hz,1h),1.13(d,j=6.8hz,6h).
13
c nmr(100mhz,cdcl3)δ138.4,136.4,135.1,132.5,132.3,128.8,127.3,127.1,126.9,126.8,125.5,123.0,55.3,21.6.
[0049]
实施例9
[0050]
将56.4mg(0.2mmol)芳基磺胺类反应底物ii
‑
4、60.3mg(0.2mmol)n
‑
bu4noac溶于6.0ml乙腈与六氟异丙醇体积比为2:1的混合溶液中,以6mm碳棒作为阳极,以1cm
×
1cm铂片作为阴极,在15ma电流作用下反应1.5小时(薄层色谱监测反应完全)后,将反应液浓缩,硅胶柱色谱分离(乙酸乙酯/石油醚=1~10,v/v)得到39.2mg产物ii
’‑
4,收率为70%;其反应方程式如下:
[0051][0052]
所得产物的结构表征数据为:1h nmr(600mhz,cdcl3)δ8.01(dd,j=7.8,1.2hz,1h),7.94(d,j=8.4hz,1h),7.92(d,j=7.8hz,1h),7.72(td,j=7.8,1.2hz,1h),7.59(td,j=7.2,0.6hz,,1h),7.32
–
7.29(m,2h),3.46(s,3h);
13
c nmr(100mhz,cdcl3)δ140.2,136.0,133.9,132.6,131.5,128.5,126.6,125.3,124.7,122.4,122.2,119.0,32.2.
[0053]
实施例10
[0054]
将68.7mg(0.2mmol)芳基磺胺类反应底物ii
‑
5、60.3mg(0.2mmol)n
‑
bu4noac溶于6.0ml乙腈与六氟异丙醇体积比为2:1的混合溶液中,以6mm碳棒作为阳极,以1cm
×
1cm铂片作为阴极,在15ma电流作用下反应1.5小时(薄层色谱监测反应完全)后,将反应液浓缩,硅胶柱色谱分离(乙酸乙酯/石油醚=1~10,v/v)得到39.2mg产物ii
’‑
5,收率为66%;其反应方程式如下:
[0055]
[0056]
所得产物的结构表征数据为:1h nmr(400mhz,cdcl3)δ8.08(d,j=8.0hz,1h),8.00(dd,j=7.6,1.2hz,1h),7.94(d,j=8.0hz,1h),7.74(td,j=7.6,1.2hz,1h),7.69
–
7.62(m,3h),4.47(hept,j=6.8hz,1h),1.14(d,j=6.8hz,6h);
13
c nmr(150mhz,cdcl3)δ137.71,136.92,132.65,131.79,131.69,131.57,131.35,129.61,126.36,126.03,124.23,124.0(q=3.8hz),123.6(q=3.4hz),123.15,122.43,55.4,21.6.
[0057]
实施例11
[0058]
将57.9mg(0.2mmol)芳基磺胺类反应底物iii
‑
1、60.3mg(0.2mmol)n
‑
bu4noac溶于6.0ml乙腈与六氟异丙醇体积比为2:1的混合溶液中,以6mm碳棒作为阳极,以1cm
×
1cm铂片作为阴极,在15ma电流作用下反应1.5小时(薄层色谱监测反应完全)后,将反应液浓缩,硅胶柱色谱分离(乙酸乙酯/石油醚=1~10,v/v)得到23.0mg产物iii
’‑
1,收率为40%;其反应方程式如下:
[0059][0060]
所得产物的结构表征数据为:1h nmr(400mhz,cdcl3)δ8.03(d,j=7.8hz,1h),7.68(t,j=7.6hz,1h),7.60(d,j=7.6hz,1h),7.54(t,j=7.6hz,1h),7.47(s,2h),7.42(s,2h),4.54
–
4.40(m,1h),3.84(s,2h),1.23(d,j=6.4hz,6h),见图3;
13
c nmr(150mhz,cdcl3)δ140.8,139.2,138.0,134.1,132.8,130.1,129.6,129.2,129.02,128.4,128.3,126.2,51.0,45.4,19.8.
[0061]
实施例12
[0062]
将60.7mg(0.2mmol)芳基磺胺类反应底物iii
‑
2、60.3mg(0.2mmol)n
‑
bu4noac溶于6.0ml乙腈与六氟异丙醇体积比为2:1的混合溶液中,以6mm碳棒作为阳极,以1cm
×
1cm铂片作为阴极,在15ma电流作用下反应1.5小时(薄层色谱监测反应完全)后,将反应液浓缩,硅胶柱色谱分离(乙酸乙酯/石油醚=1~10,v/v)得到24.1mg产物iii
’‑
2,收率为41%;其反应方程式如下:
[0063][0064]
所得产物的结构表征数据为:1h nmr(400mhz,cdcl3)δ8.11(dd,j=8.0,1.2hz,1h),7.68(td,j=7.6,1.2hz,1h),7.60(dd,j=7.6,1.2hz,1h),7.55
–
7.51(m,2h),7.46(td,j=7.2,1.2hz,1h),7.40(td,j=7.2,1.6hz,1h),7.31(dd,j=7.2,1.2hz,1h),4.66
–
4.53(m,1h),4.48(q,j=6.8hz,1h),1.40(d,j=6.8hz,3h),1.33(d,j=6.8hz,3h),0.96(d,j=6.8hz,3h);
13
c nmr(150mhz,cdcl3)δ140.9,140.1,139.1,138.5,133.0,129.7,
129.6,129.2,128.8,128.0,126.7,55.4,49.2,22.4,21.9,21.5;dept(100mhz,cdcl3)δ132.8,129.5,129.4,129.0,128.6,127.8,126.5,55.2,49.0,22.2,21.7,21.2。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1