一种2,2`-双琥珀酰亚胺衍生物的电化学合成方法
一种2,2'-双琥珀酰亚胺衍生物的电化学合成方法
技术领域
1.本发明属于电化学合成技术领域,具体涉及一种2,2'-双琥珀酰亚胺衍生物的电化学合成方法。
背景技术:
2.琥珀酰亚胺具有独特的五元环状结构,2,2'-双琥珀酰亚胺衍生物在材料生产加工中有很多应用。例如:n,n-双甲基-2,2'-双琥珀酰亚胺可做为反应中间体用于制备合成多孔硅铝酸盐emm-37所需的结构导向剂。2009年,秋山等报道,含有n,n-双(3-羟基苯基)-2,2'-双琥珀酰亚胺的硬化剂能够使其制备的环氧树脂提高玻璃化转变温度,使弹性模量保持在较低水平,所制备的环氧树脂内应力低,可用于封装半导体器件。此外,2,2'-双琥珀酰亚胺结构在异氰酸盐的生产、光敏聚合物的合成、液晶对准膜的制造、液晶对准剂的制造、液晶取向膜的制备 、光导纤维的制造等方面也有很多报道,相应结构片段的加入能使高分子聚合物具有更优异的性质。
3.目前,该类化合物的传统合成方法主要是用1,2,3,4-丁烷四羧酸二酐与胺反应。例如日本日产化学株式会社曾报道,将间氨基苯酚溶于二甲苯,向其中滴加溶于dmf的1,2,3,4-丁烷四羧酸二酐溶液,室温下反应2小时,再加入甲苯与甲苯磺酸,在140℃反应4小时,脱水闭环,得到n,n-双(3-羟基苯基)-2,2'-双琥珀酰亚胺。传统化学合成方法一般需要分步进行,1,2,3,4-丁烷四羧酸二酐的价格也较昂贵,反应需在高温下进行,条件较为苛刻。而2,2'-双琥珀酰亚胺衍生物具有广泛的应用前景,开发绿色、简便的合成方法对于2,2'-双琥珀酰亚胺衍生物的发展具有重要意义。
技术实现要素:
4.本发明的目的在于针对上述现有技术的不足之处,提供一种2,2'-双琥珀酰亚胺衍生物的电化学合成方法。
5.为解决上述技术问题,本发明采用如下技术方案:一种2,2'-双琥珀酰亚胺衍生物的电化学合成方法,以溶解有马来酰亚胺衍生物的有机溶剂为电解反应原料,在电解条件下合成2,2'-双琥珀酰亚胺衍生物;所述马来酰亚胺衍生物的结构式如式(ⅰ)所示:所述2,2'-双琥珀酰亚胺衍生物的结构式如式(ⅱ)所示:
其中,r为氢原子、取代或未取代的脂肪族、取代或未取代的芳香族中的任意一种。
6.优选地,所述电解条件为:阳极电极为铂电极、石墨电极中的任意一种;阴极电极为铂电极、石墨电极中的任意一种。
7.优选地,所述电解条件为:电流密度为1.25~20 ma∕cm2;电解反应温度为20~50 ℃;电解反应时间为10~30 h。
8.优选地,所述有机溶剂为dmf、乙醇、乙腈、四氢呋喃、六氟异丙醇中的任意一种。
9.优选地,所述电解反应原料中,还加入有支持电解质,所述支持电解质包括nano3。
10.优选地,所述支持电解质还包括季铵盐。
11.优选地,所述电解反应原料中,还加入有ph值为3.00~10.00的ph缓冲液。
12.优选地,所述ph缓冲液为磷酸缓冲液、醋酸-醋酸钠缓冲液,磷酸氢二钠-柠檬酸缓冲液中的任意一种。
13.优选地,所述有机溶剂与所述ph缓冲液的体积比为(0.1~10):1。
14.优选地,电解反应合成的所述2,2'-双琥珀酰亚胺衍生物经过以下纯化过程处理:将含有所述2,2'-双琥珀酰亚胺衍生物的电解液倒入水中,析出产物,过滤洗涤并干燥。
15.与现有技术相比,本发明的有益效果为:本发明的一种2,2'-双琥珀酰亚胺衍生物的电化学合成方法克服了传统的2,2'-双琥珀酰亚胺的制备方法需要的原料1,2,3,4-丁烷四羧酸二酐的价格昂贵,且反应需在高温下进行、条件苛刻的缺陷;相对于传统的有机合成,用电化学法合成2,2'-双琥珀酰亚胺衍生物具有显著的优势,电化学反应是通过反应物在电极上得失电子实现的,不用加入额外的还原剂或金属催化剂,减少了物质消耗,从而减少了环境污染;反应在室温常压下即可进行,反应条件温和,后处理简单,这对节约能源、降低设备投资十分有利;工艺流程简单,操作简便,反应容易控制,一锅反应即可得到2,2'-双琥珀酰亚胺衍生物;反应原料廉价易得,且使用时十分安全;反应过程绿色友好,符合绿色化学工艺的要求。
16.本发明的附加优点、目的以及特征将在下面的描述中将部分地加以阐述,且将对于本领域普通技术人员在研究下文后部分地变得明显,或者可以根据本发明的实践而获知。
17.本领域技术人员将会理解的是,能够用本发明实现的目的和优点不限于以上具体所述,并且根据以下详细说明将更清楚地理解本发明能够实现的上述和其他目的。
附图说明
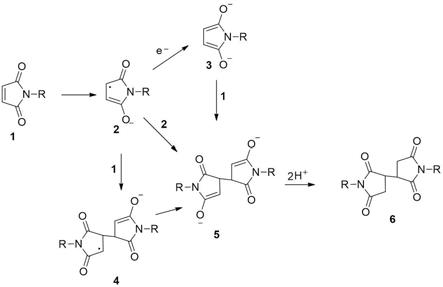
18.图1是本发明实施例提供的一种2,2'-双琥珀酰亚胺衍生物的电化学合成方法中可能的反应机理示意图;
图2是本发明实施例1的产物的氢谱图;图3是本发明实施例1的产物的碳谱图;图4是本发明实施例7的产物的氢谱图;图5是本发明实施例7的产物的碳谱图;图6是本发明实施例11的产物的氢谱图;图7是本发明实施例11的产物的碳谱图;图8是本发明实施例13的产物的氢谱图;图9是本发明实施例13的产物的碳谱图;图10是本发明实施例15的产物的氢谱图;图11是本发明实施例15的产物的碳谱图;图12是本发明实施例15的产物的
13
c-dept 135谱图。
具体实施方式
19.以下通过特定的具体实例说明本发明的实施方式,本领域技术人员可由本说明书所揭露的内容轻易地了解本发明的其他优点与功效。本发明还可以通过另外不同的具体实施方式加以实施或应用,本说明书中的各项细节也可以基于不同观点与应用,在没有背离本发明的精神下进行各种修饰或改变。
20.须知,下列实施例中未具体注明的工艺设备或装置均采用本领域内的常规设备或装置。
21.此外应理解,本发明中提到的一个或多个方法步骤并不排斥在所述组合步骤前后还可以存在其他方法步骤或在这些明确提到的步骤之间还可以插入其他方法步骤,除非另有说明;而且,除非另有说明,各方法步骤的编号仅为鉴别各方法步骤的便利工具,而非为限制各方法步骤的排列次序或限定本发明可实施的范围,其相对关系的改变或调整,在无实质变更技术内容的情况下,当亦视为本发明可实施的范畴。
22.本发明实施例提供一种一锅完成、绿色友好、简单高效的2,2'-双琥珀酰亚胺衍生物的电化学合成方法,该方法通过在一室电解池内加入溶解有马来酰亚胺衍生物的有机溶剂为电解反应原料,在电解条件下合成2,2'-双琥珀酰亚胺衍生物;马来酰亚胺衍生物的结构式如式(ⅰ)所示:2,2'-双琥珀酰亚胺衍生物的结构式如式(ⅱ)所示:
其中,r为氢原子、取代或未取代的脂肪族、取代或未取代的芳香族中的任意一种。
23.在电解反应过程中,采用薄层色谱(tlc)监控反应的进程,反应结束后,通过以下纯化过程处理:将含有2,2'-双琥珀酰亚胺衍生物的电解液倒入水中,析出固体,过滤洗涤干燥,得到产物2,2'-双琥珀酰亚胺衍生物。
24.为避免电极参与氧化还原反应,干扰目标反应,电极材质选用惰性电极,优选地,电解条件为:阳极电极为铂电极、石墨电极中的任意一种;阴极电极为铂电极、石墨电极中的任意一种。更优选地,阳极电极为铂电极,阴极电极为铂电极。
25.优选地,电解条件控制为:恒电流电解,电流密度为1.25~20 ma∕cm2;电解反应温度为20~50 ℃;电解反应时间为10~30 h。电流的大小对反应有一定的影响,如果电流过大,会导致底物或产物被过分氧化,而电流过小,则会导致反应无法开始。提高电解反应温度对降低过电位,提高电流密度有益;但是过高的电解反应温度会使副反应增多,使副反应速率同时增大,同时可能会使产物分解。长时间的反应虽然能最大程度提高反应的转化率,但后期的产率变化提升缓慢,且能耗较高,综合产率与成本的考虑,更优选地,电解反应时间为10~24 h。
26.优选地,有机溶剂为dmf、乙醇、乙腈、四氢呋喃、六氟异丙醇中的任意一种。
27.优选地,电解反应原料中,还加入有支持电解质,该支持电解质包括nano3。更优选地,该支持电解质还包括季铵盐,如四丁基四氟硼酸铵、四丁基硝酸铵、四丁基硫酸氢铵、四丁基溴化铵、四丁基碘化铵、四丁基高氯酸铵等。季铵盐可以作为电解质增强电解液的电导率,增加反应原料的溶解度。
28.优选地,电解反应原料中,还加入有ph值为3.00~10.00的ph缓冲液。该ph缓冲液可选为磷酸缓冲液、醋酸-醋酸钠缓冲液,磷酸氢二钠-柠檬酸缓冲液中的任意一种。更优选地,ph缓冲液ph值为6.00~8.00。
29.优选地,有机溶剂与ph缓冲液的体积比可为(0.1~10):1。
30.本发明的合成方法使用马来酰亚胺衍生物为起始原料,在一室电解池内,恒电流电解,一锅法得到2,2'-双琥珀酰亚胺衍生物,操作方便,后处理简单,反应条件温和,原料便宜易得且使用时十分安全,可以有效地降低生产成本;反应过程绿色友好,符合绿色化学工艺的要求。本发明的合成方法克服了传统的2,2'-双琥珀酰亚胺的制备方法需要的原料1,2,3,4-丁烷四羧酸二酐的价格昂贵,且反应需在高温下进行、条件苛刻的缺陷;相对于传统的有机合成,用电化学法合成2,2'-双琥珀酰亚胺衍生物具有显著的优势,电化学反应是通过反应物在电极上得失电子实现的,不用加入额外的还原剂或金属催化剂,减少了物质消耗,从而减少了环境污染;反应在室温常压下即可进行,反应条件温和,后处理简单,这对节约能源、降低设备投资十分有利;工艺流程简单,操作简便,反应容易控制。
31.本发明的一种2,2'-双琥珀酰亚胺衍生物的电化学合成方法中可能的反应机理如图1所示:马来酰亚胺衍生物的碳碳双键两侧分别接有一个羰基,羰基的吸电子作用使碳碳双键容易接收亲核类试剂的进攻,发生迈克尔类型的加成反应。电解反应时,马来酰亚胺衍生物首先通过得到一个电子形成自由基负离子(1至2),继而自由基负离子2发生自由基-自由基二聚(2至5)或自由基-底物加成反应(2至4)或负离子-底物加成反应(3至5),进一步在溶剂中捕获氢离子得到产物2,2'-双琥珀酰亚胺衍生物(6)。
32.以下结合具体实施例做进一步说明。
33.实施例1n,n'-二苄基-2,2'-双琥珀酰亚胺的合成方法如下:恒电流电解,电流密度为20 ma∕cm2,铂片(2cm
×
2cm)为阳极电极与阴极电极,在一室电解池内,加入底物n-苄基-马来酰亚胺(1 mmol),加入nano3(10 mmol)为电解质,加入dmf(5 ml)为有机溶剂,加入ph(6~7)的磷酸缓冲液(15 ml),40℃水浴,以薄层色谱法追踪反应进程,反应时间20 h。反应结束后,将反应液倒入300 ml水中,析出白色固体,过滤干燥,收率为70%。
34.n,n'-二苄基-2,2'-双琥珀酰亚胺结构式为。
35.实施例2~实施例15以不同马来酰亚胺衍生物为反应原料合成对应的2,2'-双琥珀酰亚胺衍生物,实验参数与实验步骤与实施例1相同。实施例实验结果见表1。
36.实施例16-实施例21用不同有机溶剂合成n,n'-二苄基-2,2'-双琥珀酰亚胺:恒电流电解,电流密度为5ma∕cm2,铂片(2cm
×
2cm)为阳极电极与阴极电极,在一室电解池内,加入底物n-苄基-马来酰亚胺(1 mmol),加入nano3(6 mol)为电解质,加入5 ml有机溶剂,加入ph(6~7)的磷酸缓冲液(15 ml),20℃水浴,以薄层色谱法追踪反应进程,反应时间15 h。反应结束后,将反应液倒入300 ml水中,析出白色固体,过滤干燥。实施例实验结果见表2。
37.比较实施例16、实施例17、实施例18、实施例19、实施例20和实施例21,本发明选用了和水溶液互溶程度较好的几种溶剂:dmf、乙醇、四氢呋喃、异丙醇、六氟异丙醇、乙腈进行比较,可以看出有机溶剂对反应的影响较大,发现以dmf为有机溶剂时,反应产率较高。
38.实施例22~实施例25使用不同反应时间合成n,n'-二苄基-2,2'-双琥珀酰亚胺:恒电流电解,电流密度为5ma∕cm2,铂片(2cm
×
2cm)为阳极电极与阴极电极,在一室电解池内,加入底物n-苄基-马来酰亚胺(1 mmol),加入nano3(6 mol)为电解质,加入dmf(5 ml)为有机溶剂,加入ph(6~7)的磷酸缓冲液(15 ml),并加入四丁基溴化铵 (0.05mmol),20℃水浴,以薄层色谱法追踪反应进程。反应结束后,将反应液倒入300 ml水中,析出白色固体,过滤干燥。实施例实验结果见表3。
39.比较实施例22、实施例23、实施例24、实施例25,反应时间的长短也会对反应产率有所影响,反应时间过短,可能导致反应物反应不完全,而反应时间过长,则可能导致电解过度,比较反应时间为10 h,15 h,20 h,27 h的电解实验,在其他条件相同的情况,反应时间为20 h时产率较高。
40.实施例26~实施例36使用不同支持电解质合成n,n'-二苄基-2,2'-双琥珀酰亚胺:恒电流电解,电流密度为5ma∕cm2,铂片(2cm
×
2cm)为阳极电极与阴极电极,在一室电解池内,加入底物n-苄基-马来酰亚胺(1 mmol),加入nano3(6 mol)为电解质,加入dmf(5 ml)为有机溶剂,加入ph(6~7)的磷酸缓冲液(15 ml),40℃水浴,以薄层色谱法追踪反应进程,反应时间20 h。反应结束后,将反应液倒入300 ml水中,析出白色固体,过滤干燥。实施例实验结果见表4。
41.比较实施例26、实施例27、实施例28、实施例29、实施例30、实施例31、实施例32、实施例33、实施例34、实施例35、实施例36,季铵盐可以作为电解质增强电解液的电导率,增加原料的溶解度。在n-苄基-马来酰亚胺的电解二聚实验中,除了加入nano3作为支持电解质,还加入季铵盐,分别选用四丁基四氟硼酸铵、四丁基硝酸铵、四丁基硫酸氢铵、四丁基溴化铵、四丁基碘化铵、四丁基高氯酸铵作为额外的支持电解质,并调整四丁基溴化铵的量进行实验,发现加入季铵盐并没有使产率有较大提升,甚至加入某些季铵盐会使反应产率有所下降,本发明仍选择只加入nano3为支持电解质。
42.实施例37~实施例38使用不同反应电极合成n,n'-二苄基-2,2'-双琥珀酰亚胺:恒电流电解,电流密度为5ma∕cm2,在一室电解池内,加入底物n-苄基-马来酰亚胺(1 mmol),加入nano3(6 mol)为电解质,加入dmf(5 ml)为有机溶剂,加入ph(6~7)的磷酸缓冲液(15 ml),40℃水浴,以薄层色谱法追踪反应进程,反应时间20 h。反应结束后,将反应液倒入300 ml水中,析出白色固体,过滤干燥。实施例实验结果见表5。
43.比较实施例26、实施例37、实施例38,电极的搭配对反应非常重要,本发明尝试了铂电极(阳极)-铂电极(阴极),石磨棒(阳极)-铂电极(阴极),铂电极(阳极)-石磨棒(阴极)组合进行反应。实验显示,铂电极(阳极)-铂电极(阴极)的组合作为阴极与阳极,反应产率更高。铂电极具有化学性质稳定,氧过电位大,且容易加工的特点,经常作为有机物或无机物电解氧化的研究电极,且铂板电极与反应液的接触面积更大,反应速率更快,石墨电极具有多孔性,使用时会因浸入电解液或者氧气而影响反应。
44.实施例39~实施例41使用不同电流密度合成n,n'-二苄基-2,2'-双琥珀酰亚胺:恒电流电解,在一室电解池内,加入底物n-苄基-马来酰亚胺(1 mmol),加入nano3(6 mol)为电解质,加入dmf(5 ml)为有机溶剂,加入ph(6~7)的磷酸缓冲液(15 ml),40℃水浴,以薄层色谱法追踪反应进程,反应时间20 h。反应结束后,将反应液倒入300 ml水中,析出白色固体,过滤干燥。实施例实验结果见表6。
45.比较实施例26、实施例39、实施例40、实施例41,电流密度的大小对反应有一定的影响,如果电流过大,副产物可能增多,而电流过小,则会导致反应不完全。
46.实施例42~实施例43使用不同反应温度合成n,n'-二苄基-2,2'-双琥珀酰亚胺:恒电流电解,在一室电解池内,铂片(2cm
×
2cm)为阳极电极与阴极电极,加入底物n-苄基-马来酰亚胺(1 mmol),加入nano3(6 mol)为电解质,加入dmf(5 ml)为有机溶剂,加入ph(6~7)的磷酸缓冲液(15 ml),以薄层色谱法追踪反应进程,反应时间20 h。反应结束后,将反应液倒入300 ml水中,析出白色固体,过滤干燥。实施例实验结果见表7。
47.比较实施例42、实施例26、实施例43,提高温度对于提高电流密度、降低过电位有益,但是温度过高会使某些副反应加速,产物可能分解,实验中,发现反应在40℃水浴中,产率较高,而在35℃水浴中有原料未反应完。
48.实施例44~实施例45使用不同量的nano3合成n,n'-二苄基-2,2'-双琥珀酰亚胺:恒电流电解,在一室电解池内,铂片(2cm
×
2cm)为阳极电极与阴极电极,加入底物n-苄基-马来酰亚胺(1 mmol),加入nano3为电解质,加入dmf(5 ml)为有机溶剂,加入ph(6~7)的磷酸缓冲液(15 ml),40℃水浴,以薄层色谱法追踪反应进程,反应时间20 h。反应结束后,将反应液倒入300 ml水中,析出白色固体,过滤干燥。实施例实验结果见表8。
49.比较实施例26、实施例1、实施例44,实施例45,nano3作为支持电解质,nano3的量可以影响反应体系的导电性,尽管磷酸缓冲液也具有一定的导电性,但是仍需要筛选合适的电解质,以确定合适的nano3的量。经过比较,确定10 mmol 的nano3对反应来说最合适。
50.实施例46~实施例47使用不同ph的磷酸缓冲液合成n,n'-二苄基-2,2'-双琥珀酰亚胺:恒电流电解,在一室电解池内,铂片(2cm
×
2cm)为阳极电极与阴极电极,加入底物n-苄基-马来酰亚胺(1 mmol),加入nano3(10 mol)为电解质,加入dmf(5 ml)为有机溶剂,加入磷酸缓冲液(15 ml),40℃水浴,以薄层色谱法追踪反应进程,反应时间20 h。反应结束后,将反应液倒入300 ml水中,析出白色固体,过滤干燥。实施例实验结果见表9。
51.比较实施例46、实施例1、实施例47,在低ph条件下,n-苄基-马来酰亚胺在自由基阴离子质子化后容易被快速还为琥珀酰亚胺,而n-苄基-马来酰亚胺在碱性条件下易水解,因此偏中性的ph 6~7的磷酸缓冲液环境更加有利于n-苄基-马来酰亚胺的加氢二聚反应。
52.实施例48~实施例49使用不同体积比的dmf与磷酸缓冲液混合液,合成n,n'-二苄基-2,2'-双琥珀酰亚胺:恒电流电解,在一室电解池内,铂片(2cm
×
2cm)为阳极电极与阴极电极,加入底物n-苄基-马来酰亚胺(1 mmol),加入nano3(10 mol)为电解质,加入使用不同体积比的dmf与ph(6~7)磷酸缓冲液混合液(反应液总体积为20 ml),加入磷酸缓冲液(15 ml),40℃水浴,以薄层色谱法追踪反应进程,反应时间20 h。反应结束后,将反应液倒入300 ml水中,析出白色固体,过滤干燥。实施例实验结果见表10。
53.比较实施例48、实施例1、实施例49,不同体积比的dmf与磷酸缓冲液下,都可以合成n,n'-二苄基-2,2'-双琥珀酰亚胺,但是体积比不同,底物溶解度也不同,会影响电导率,针对n-苄基-马来酰亚胺,体积比为5:15可以得到较高产率的n,n'-二苄基-2,2'-双琥珀酰亚胺。
54.实施例50恒电流电解,电流密度为20ma∕cm2,铂片(2cm
×
2cm)为阳极电极与阴极电极,在一室电解池内,加入底物n-苄基-马来酰亚胺(1mmol),加入nano3(10mmol)为电解质,加入dmf(5ml)为有机溶剂,加入ph(6~7)的醋酸-醋酸钠缓冲液(15ml),40℃水浴,以薄层色谱法追踪反应进程,反应时间20h。反应结束后,将反应液倒入300ml水中,析出白色固体,过滤干燥,收率为58%。
55.实施例51恒电流电解,电流密度为20ma∕cm2,铂片(2cm
×
2cm)为阳极电极与阴极电极,在一室电解池内,加入底物n-苄基-马来酰亚胺(1mmol),加入nano3(10mmol)为电解质,加入dmf(5ml)为有机溶剂,加入ph(4~5)的磷酸氢二钠-柠檬酸缓冲液(15ml),40℃水浴,以薄层色谱法追踪反应进程,反应时间20h。反应结束后,将反应液倒入300ml水中,析出白色固体,过滤干燥,收率为55%。
56.实施例中的产物结构表征:核磁(1hnmr,
13
cnmr,
13
c-dept135nmr):brukeravanceⅲ500mhz;质谱:agilent6210toflc/ms;熔点:上海庚雨有限公司,显微熔点仪,x-4a;红外光谱:brukertensor27。
57.实施例1中产物的氢谱、碳谱分别如图2、图3所示,表征数据如下:whitesolid,m.p.192-193
º
c;1hnmr(500mhz,cdcl3):δ7.31-7.28(m,10h),4.63(d,j=14.10hz,2h),4.56(d,j=14.15hz,2h),3.36-3.32(m,2h),2.85(dd,j1=9.15hz,j2=18.20hz,2h),2.64(dd,j1=4.90hz,j2=18.15hz,2h);
13
cnmr(125mhz,cdcl3):δ176.89,174.59,135.37,128.74,128.64,128.14,42.65,39.56,31.81;hrms(esi+)calculatedforc
22h20
n2o4[m+na]
+
399.1315,found399.1284;irν
max
(cm-1
):2929,1697,1427,1401,1336,1167,928,742,698.实施例2中产物的氢谱、碳谱表征数据如下:whitesolid,m.p.83-85
º
c;1hnmr(500mhz,cdcl3):δ3.44(t,j=7.45hz,4h),3.33-3.29(m,2h),2.85(dd,j1=9.15hz,j2=18.10hz,2h),2.65(dd,j1=5.15hz,j2=18.10hz,2h),1.50-1.48(m,4h),1.28-1.22(m,12h),0.85(t,j=6.65hz,6h);
13
cnmr(125mhz,cdcl3):δ177.28,175.02,39.36,39.00,31.79,31.15,27.46,26.28,22.35,13.85;hrms(esi+)calculatedforc
20h32
n2o4[m+h]
+
364.2435,found365.2434;irν
max
(cm-1
):2958,2933,2858,1697,1440,1410,1346,1129.实施例3中产物的氢谱、碳谱表征数据如下:whitesolid,m.p.134-135
º
c;1hnmr(500mhz,cdcl3):δ4.30(sep,j=7.05hz,2h),3.21-3.17(m,2h),2.80(dd,j1=9.35hz,j2=18.15hz,2h),
4h), 7.44-7.41 (m, 2h), 7.28-7.25 (m, 4h), 3.60-3.56 (m, 2h), 3.18 (dd, j
1 = 9.20 hz, j
2 = 18.10 hz, 2h), 3.09 (dd, j
1 = 5.75 hz, j
2 = 18.05 hz, 2h); 13
c nmr (125 mhz, cdcl3): δ 176.60, 173.98, 131.45, 129.30, 128.99, 126.44, 40.03, 32.91; hrms (esi+) calculated for c
20h16
n2o
4 [m + na]
+ 371.1002, found 371.1017; irν
max (cm-1
): 2923, 1707, 1502, 1389, 1194, 760, 698.实施例9中产物的氢谱、碳谱表征数据如下:white solid, m.p. 242-43
º
c; 1
h nmr (500 mhz, cdcl3): δ 7.25-7.23 (m, 4h), 7.19-7.15 (m, 4h), 3.56-3.52 (m, 2h), 3.20 (dd, j
1 = 9.15 hz, j
2 = 18.20 hz, 2h), 3.13 (dd, j
1 = 5.95 hz, j
2 = 18.15 hz, 2h); 13
c nmr (125 mhz, cdcl3): δ 176.59, 173.80, 162.46 (d, j = 248.06 hz), 128.32 (d, j = 8.71 hz), 127.84 (d, j = 3.36 hz), 116.39 (d, j = 22.93 hz), 40.10, 33.26; hrms (esi+) calculated for c
20h14
f2n2o
4 [m + na]
+ 403.0814, found 407.0818; irν
max
(cm-1
): 2977, 2949, 1709, 1694, 1513, 1396, 1234, 1191, 1175, 845.实施例10中产物的氢谱、碳谱表征数据如下:light yellow liquid; 1
h nmr (500 mhz, cdcl3): δ 4.25-4.17 (m, 4h), 3.75 (t, j = 5.05 hz, 4h), 3.46-3.42 (m, 2h), 2.87 (dd, j
1 = 9.10 hz, j
2 = 18.20 hz, 2h), 2.65 (dd, j
1 = 5.25 hz, j
2 = 18.25 hz, 2h), 2.00 (s, 6h); 13
c nmr (125 mhz, cdcl3): δ 177.07, 174.81, 170.71, 60.46, 39.18, 38.06, 30.93, 20.52; hrms (esi+) calculated for c
16h20
n2o
8 [m + na]
+ 391.1112, found 391.1125; irν
max (cm-1
): 2962, 1739, 1702, 1401,1371, 1335, 1235, 1192, 1115, 1058.实施例11中产物的氢谱、碳谱分别如图6、图7所示,表征数据如下:white solid, m.p. 190-191
º
c; 1
h nmr (500 mhz, cdcl3): δ 7.44-7.37 (m, 4h), 7.30-7.29 (m, 2h), 7.20-7.15 (m, 2h), 3.55-3.51 (m, 2h), 3.19 (dd, j
1 = 9.05 hz, j
2 = 18.20 hz, 2h), 3.13 (dd, j
1 = 6.10 hz, j
2 = 18.15 hz, 2h); 13
c nmr (125 mhz, cdcl3): δ 176.26, 173.44, 134.87, 132.41, 130.25, 129.22, 126.69, 124.60, 40.08, 33.23; hrms (esi+) calculated for c
20h14
cl2n2o
4 [m + na]
+ 439.0222, found 439.0236; irν
max (cm-1
): 3072, 2951, 1706, 1593, 1482, 1389, 1186, 785, 687.实施例12中产物的氢谱、碳谱表征数据如下:white solid, m.p. 235-236
º
c; 1
h nmr (500 mhz, cdcl3): δ 7.39-7.27 (m, 6h), 7.08-6.99 (m, 2h), 3.63-3.57 (m, 2h), 3.29-3.13 (m, 4h), 2.18 (s, 2.61h), 2.11 (s, 2.04h), 2.09 (s, 1.52h); 13
c nmr (125 mhz, cdcl3): δ 176.73, 176.67, 176.59, 174.04, 174.00, 173.97, 173.94, 136.07, 136.02, 135.37, 131.38, 131.35, 131.16, 130.69, 130.65, 129.85, 129.81, 128.12, 128.09, 127.76, 127.12, 127.08, 127.03, 127.00,40.69, 40.28, 40.10, 39.66, 33.59, 33.44, 33.28, 33.23, 17.75, 17.73, 17.67, 17.65; hrms (esi+) calculated for c
22h20
n2o
4 [m + h]
+ 377.1130, found 377.1116; irν
max
(cm-1
): 3063, 3033, 2992, 2932, 1704, 1494, 1461, 1387, 1188, 925, 851, 755, 717, 659, 628.实施例13中产物的氢谱、碳谱分别如图8、图9所示,表征数据如下:
white solid, m.p. 191-193
º
c; 1
h nmr (500 mhz, cdcl3): δ 7.39-7.35 (m, 2h), 7.25-7.23 (m, 2h), 7.12-7.03 (m, 4h), 3.58-3.54 (m, 2h), 3.16 (dd, j
1 = 9.20 hz, j
2 = 18.15 hz, 2h), 3.07 (dd, j
1 = 5.65 hz, j
2 = 18.15 hz, 2h), 2.39 (s, 6h); 13
c nmr (125 mhz, cdcl3): δ 176.69, 174.13, 139.43, 131.32, 129.88, 129.12, 127.06, 127.01, 123.55, 40.04, 32.87, 21.32; hrms (esi+) calculated for c
22h20
n2o
4 [m + h]
+ 377.1497, found 377.1487; irν
max
(cm-1
): 2924, 1710, 1491, 1385, 1186, 696.实施例14中产物的氢谱、碳谱表征数据如下:white solid, m.p. 260-261
º
c; 1
h nmr (500 mhz, cdcl3): δ 7.31-7.28 (m, 4h), 7.23-7.20 (m, 4h), 3.57-3.53 (m, 2h), 3.18 (dd, j
1 = 9.25 hz, j
2 = 18.10 hz, 2h), 3.10 (dd, j
1 = 5.95 hz, j
2 = 18.05 hz, 2h), 2.32 (s, 6h); 13
c nmr (125 mhz, dmso+ cdcl3): δ 175.64, 173.14, 167.15, 148.47, 127.83, 126.27, 120.59, 38.10, 30.51, 19.16; hrms (esi+) calculated for c
24h20
n2o
8 [m + na]
+ 487.1112, found 487.1076; irν
max
(cm-1
): 2976, 2950, 1753, 1705, 1693, 1512, 1394, 1197, 1182, 1171.实施例15中产物的氢谱、碳谱、13c-dept 135谱分别如图10、图11、图12所示,表征数据如下:white solid, m.p. 293-295
º
c; 1
h nmr (500 mhz, dmso): δ 9.73 (s, 2h), 7.02-6.99 (m, 4h), 6.85-6.82 (m, 4h), 3.66-3.62 (m, 2h), 2.95 (dd, j
1 = 8.95 hz, j
2 = 17.90 hz, 4h), 2.87 (dd, j
1 = 5.35 hz, j
2 = 17.90 hz, 2h); 13
c nmr (125 mhz, dmso): δ 177.75, 175.42, 157.33, 128.20, 123.32, 115.39, 39.57, 31.61; hrms (esi+) calculated for c
20h16
n2o
6 [m + na]
+ 403.0901, found 403.0910; irν
max
(cm-1
): 3392, 2924, 1700, 1518, 1397, 1245, 1186.本发明的保护范围不限于上述的实施例,显然,本领域的技术人员可以对本发明进行各种改动和变形而不脱离本发明的范围和精神。倘若这些改动和变形属于本发明权利要求及其等同技术的范围,则本发明的意图也包含这些改动和变形在内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1