一种硼氮共配位铜单原子催化剂的制备方法及其应用
1.本发明涉及金属单原子催化剂技术领域,具体地说涉及一种硼氮共配位铜单原子催化剂的制备方法及其应用。
背景技术:
2.当前co2的转化方法颇多,其中电化学还原方法由于反应条件温和(常温、常压)、可控性良好(通过控制电极反应电位可直接控制电极表面的反应活化能)、能量来源广泛(还原过程所需电能可来自太阳能、风能、地热能和潮汐能等可再生能源)以及环境友好等优点而备受关注。
3.在co2电化学还原反应中,金属纳米催化剂相较于块体电极材料由于有效活性中心多、比表面积大、表面能高且易通过对其结构的调控实现其物性的变化等诸多特点被广泛研究。由于只有催化剂表面原子能够参与催化反应,其原子利用率较低;催化剂表面配位不饱和原子比例较少,限制其对底物分子的活化能力;催化材料活性位点不均一,难以有效调控特定产物的选择性。单原子催化剂是金属中心直接与其他配体结构结合并原子级分散于载体表面的催化材料,近些年逐渐成为多相催化领域一个新兴的研究热点。相比于金属纳米催化剂,单原子催化剂具有活性金属组分原子利用率高、活性中心结构和组成简单且金属中心电子结构可调等优势。对此单原子催化剂的研究有望替代金属纳米催化剂实现对co2电化学还原的高活性、高选择性和高稳定性。
4.目前在co2电化学还原领域是用广泛的金属元素是铜(cu),但文献报道的cu基单原子催化剂配位结构组成受限,大多为和氮(n)或氧(o)配位的cu-n或cu-o的结构,应用于co2电化学还原时得到的产物也多为一氧化碳(co),同时催化剂的活性也很有限,难以实现在较大电流密度下稳定转化co2,在较大工作电流下,催化剂的单原子结构也容易迁移团聚为纳米颗粒,从而丧失原本单原子中心的高选择性。
技术实现要素:
5.本发明所要解决的技术问题在于提供了一种具有硼、氮(b、n)共配位结构的新型cu单原子催化剂(bcn-cu)的制备方法及应用。该cu单原子催化剂应用于co2电还原反应时,能够实现大电流下cu单原子结构保持稳定不发生团聚。同时b、n共配位cu金属中心的独特电子结构有利于co2高选择性还原为甲烷(ch4)。
6.为了解决上述技术问题,本发明采用如下技术方案:一种b、n共配位的cu单原子催化剂,包括b、n均匀掺杂的碳纳米管载体和原子级分散于其表面的cu单原子。
7.进一步地,cu原子和bcn载体质量比为1.5~2.5:95~105。在实施本发明的过程中,发明人发现,采用上述配比,可以得到b、n共同配位的cu单原子催化剂。若cu原子的负载量高于该比例,则会团聚形成cu颗粒。
8.本发明还提供上述b、n共配位的cu单原子催化剂(bcn-cu)在co2电化学还原反应中的应用。
9.本发明还提供上述b、n共配位的cu单原子催化剂(bcn-cu)的制备方法,包括以下步骤:将混合液a回流加热,冷却后得到混合液b。将混合液b旋蒸至溶剂挥发溶质结晶析出,将旋蒸所得晶体充分研磨均匀得到固体c。将固体c置于管式炉中氩气氛围下充分热解即得到b、n共配位的cu单原子催化剂(bcn-cu)。
10.进一步地,配制混合液a时,尿素/单氰胺、peg-2000(聚乙二醇,平均分子量~2000)、硼酸、三水合硝酸铜和去离子水的质量体积比为19800~20200(mg)/13800~14200(mg):1980~2020(mg):590~610(mg):95~105(mg):150~170(ml)。在如上反应物浓度配比下,可以有效保证硼源(硼酸)、氮源(尿素/单氰胺)和peg-2000充分混合形成热解前驱体,同时cu离子高度分散于前驱体结构中,最终经过热解得到b、n共配位的cu单原子催化剂(bcn-cu)。
11.进一步地,混合液a加热回流时温度控制为115~125oc,搅拌速率为700~800rpm,回流时常为11~13h。该回流条件下可以保证反应充分进行,并使金属中心铜原子和与之配位的b、n原子充分分散。
12.进一步地,对混合液a回流后冷却得到的混合液b进行旋蒸处理,处理的具体操作过程如下:先将混合液b充分超声混合均匀,转移到旋蒸所用烧瓶中,控制旋蒸仪转速和水浴温度,保温蒸发约一段时间。使用旋蒸仪进行固液分离,可以有效使混合液中的固体从溶剂中均匀析出。过于剧烈的旋蒸条件会导致析出固体组分的均匀性下降,而更长的蒸发时间会导致固体中有机物组分大量分解,影响后续热解得到的单原子催化剂结构。
13.进一步地,混合液b进行旋蒸处理时,转速控制为30~50rpm,水浴温度为40~60oc。采用该旋蒸条件可以在保证固体组分均匀性的前体下实现固液高效分离。更温和的旋蒸条件不利于溶剂的挥发,而过于剧烈的旋蒸条件会导致析出固体组分的均匀性下降。
14.进一步地,混合液b进行旋蒸处理时,保温时间为1.5~2.5h。而更长的蒸发时间会导致固体中有机物组分大量分解,影响后续热解得到的单原子催化剂结构。
15.进一步地,将混合液b旋蒸所得晶体从烧瓶壁上用药勺刮下,用研钵研磨充分研磨均匀得到固体c,该固体c是热解得到单原子催化剂的前驱体,将固体c置于管式炉中惰性气氛下热解即可得到b、n共配位的cu单原子催化剂。
16.进一步地,固体粉末c在管式炉中进行热解时,采用惰性气体氩气作为保护气,气体流速控制为95~105sccm。在该气体流速下,能将热解产生的大量气体物质快速带离体系。从而保证热解过程的稳定高效进行。
17.进一步地,固体粉末c在管式炉中进行热解时,管式炉升温程序控制如下:升温速率为4~6oc/min,热解温度为850~950oc,保温时间为5.5~6.5h。在该热解条件下,可以保证前驱体的充分热解,最终得到b、n均匀掺杂的碳纳米管载体和原子级分散于其表面的b、n共同配位的cu单原子。
18.本发明的有益效果体现在:本发明以b、n均匀掺杂的碳纳米管作为载体,高分散负载cu原子,得到一种b、n共同配位的新型cu单原子催化剂。该cu单原子催化剂应用于co2电还原反应时,能够实现大电流下cu单原子结构保持稳定不发生团聚。同时b、n共配位cu金属中心的独特电子结构有利于co2高选择性还原为ch4。
19.本发明bcn-cu催化剂的制备方法,可以得到b、n共同配位的新型cu单原子结构。制
备方法采用一步热解的方式,直接将金属盐混合有机、无机前体进行热解,相较于先得到载体再负载单原子的方法,步骤大大简化。实验所需特殊设备少,产物易于分离。
附图说明
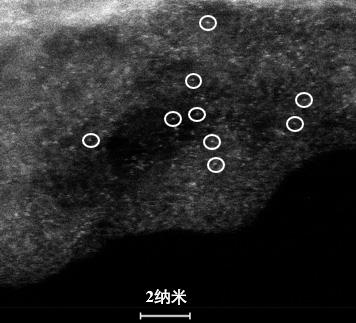
20.图1为本发明中实施例4所得b、n共同配位的cu单原子催化剂的透射电子显微镜照片;图2为本发明中实施例4所得b、n共同配位的cu单原子催化剂的扫描透射电子显微镜高角环形暗场像;图3为本发明中对实施例4所得b、n共同配位的cu单原子催化剂进行软x射线吸收测试所得硼k边吸收谱图;图4为本发明中对实施例4所得b、n共同配位的cu单原子催化剂进行软x射线吸收测试所得氮k边吸收谱图;图5为本发明中对实施例4所得b、n共同配位的cu单原子催化剂进行软x射线吸收测试所得碳k边吸收谱图;图6为本发明中对实施例4所得b、n共同配位的cu单原子催化剂中cu元素进行x射线吸收精细结构测试所得谱图;图7为本发明中实施例5流动型电解池的结构示意图;图8为本发明中实施例5所测不同电流密度下各产物的法拉第效率;图9为本发明中实施例5所测不同电流密度下甲烷法拉第效率和偏电流密度与阴极电位关系。
具体实施方式
21.下面结合实施例对本发明作进一步描述:以下实施例所使用的各种原料,如未作特别说明,均为本领域公知的市售产品。
22.实施例1bcn-cu单原子催化剂的制备(1)在250ml容积圆底烧瓶中加入20.02g尿素,2.01g peg-2000,599mg硼酸,102mg 三水合硝酸铜,170ml去离子水,放入超声机中充分混合均匀后得到混合液a。将混合液a置于油浴锅中进行加热回流,搅拌转速为725rpm,油浴锅温度设置为118oc,反应12.5h,回流反应结束后自然冷却至室温,超声均匀后得到混合液b;(2)将混合液b转移入旋蒸所用烧瓶中,进行旋蒸操作。旋蒸仪温度控制为52oc,转速设置为38rpm,旋蒸2.2h后,溶剂蒸发完全,灰色固体从混合液中充分析出。将旋蒸所用烧瓶取下,用药勺将附着在烧瓶壁上的固体刮下,并用研钵充分研磨均匀后得到固体c;(3)称取固体c 4g于氧化铝坩埚(带盖)中,并置于管式炉中进行热解操作。热解气氛为氩气,流速设置为95sccm,程序升温速率设置为5.5oc/min,热解温度设置为875oc,热解时间为6h。热解过程结束后,自然冷却至室温,将坩埚中的黑色固体研磨成粉末,即得到b、n共同配位的cu单原子催化剂(bcn-cu)。经称量,本实例中4g固体c热解共得到bcn-cu单原子催化剂127mg。
23.实施例2
bcn-cu单原子催化剂的制备(1)在250ml容积圆底烧瓶中加入19.95g尿素,2.01g peg-2000,602mg硼酸,100mg 三水合硝酸铜,180ml去离子水,放入超声机中充分混合均匀后得到混合液a。将混合液a置于油浴锅中进行加热回流,搅拌转速为750rpm,油浴锅温度设置为121oc,反应12h,回流反应结束后自然冷却至室温,超声均匀后得到混合液b;(2)将混合液b转移入旋蒸所用烧瓶中,进行旋蒸操作。旋蒸仪温度控制为49oc,转速设置为41rpm,旋蒸2h后,溶剂蒸发完全,灰色固体从混合液中充分析出。将旋蒸所用烧瓶取下,用药勺将附着在烧瓶壁上的固体刮下,并用研钵充分研磨均匀后得到固体c;(3)称取固体c 4.2g于氧化铝坩埚(带盖)中,并置于管式炉中进行热解操作。热解气氛为氩气,气体流速设置为100sccm,程序升温速率设置为5oc/min,热解温度设置为900oc,热解时间为6.5h。热解过程结束后,自然冷却至室温,将坩埚中的黑色固体研磨成粉末,即得到b、n共同配位的cu单原子催化剂(bcn-cu)。经称量,本实例中4.2 g固体c热解共得到bcn-cu单原子催化剂122mg。
24.实施例3bcn-cu单原子催化剂的制备(1)在250ml容积圆底烧瓶中加入13.85g单氰胺,1.98g peg-2000,605mg硼酸,97mg 三水合硝酸铜,170ml去离子水,放入超声机中充分混合均匀后得到混合液a。将混合液a置于油浴锅中进行加热回流,搅拌转速为800rpm,油浴锅温度设置为125oc,反应11.5h,回流反应结束后自然冷却至室温,超声均匀后得到混合液b;(2)将混合液b转移入旋蒸所用烧瓶中,进行旋蒸操作。旋蒸仪温度控制为50oc,转速设置为42rpm,旋蒸2h后,溶剂蒸发完全,深灰色固体从混合液中充分析出。将旋蒸所用烧瓶取下,用药勺将附着在烧瓶壁上的固体刮下,并用研钵充分研磨均匀后得到固体c;(3)称取固体c 3.7g于氧化铝坩埚(带盖)中,并置于管式炉中进行热解操作。热解气氛为氩气,气体流速设置为90sccm,程序升温速率设置为4.5oc/min,热解温度设置为850oc,热解时间为6.5h。热解过程结束后,自然冷却至室温,将坩埚中的黑色固体研磨成粉末,即得到b、n共同配位的cu单原子催化剂(bcn-cu)。经称量,本实例中3.7g固体c热解共得到bcn-cu单原子催化剂215mg。
25.实施例4bcn-cu单原子催化剂的制备(1)在250ml容积圆底烧瓶中加入14.01g单氰胺,2.00g peg-2000,601mg硼酸,105mg 三水合硝酸铜,150ml去离子水,放入超声机中充分混合均匀后得到混合液a。将混合液a置于油浴锅中进行加热回流,搅拌转速为750rpm,油浴锅温度设置为120oc,反应12h,回流反应结束后自然冷却至室温,超声均匀后得到混合液b;(2)将混合液b转移入旋蒸所用烧瓶中,进行旋蒸操作。旋蒸仪温度控制为55oc,转速设置为40rpm,旋蒸2.5h后,溶剂蒸发完全,深灰色固体从混合液中充分析出。将旋蒸所用烧瓶取下,用药勺将附着在烧瓶壁上的固体刮下,并用研钵充分研磨均匀后得到固体c;(3)称取固体c 3.5g于氧化铝坩埚(带盖)中,并置于管式炉中进行热解操作。热解气氛为氩气,气体流速设置为100sccm,程序升温速率设置为5oc/min,热解温度设置为900oc,热解时间为6h。热解过程结束后,自然冷却至室温,将坩埚中的黑色固体研磨成粉
末,即得到b、n共同配位的cu单原子催化剂(bcn-cu)。经称量,本实例中3.5g固体c热解共得到bcn-cu单原子催化剂204mg。
26.对实施例4所得b、n共同配位的cu单原子催化剂进行表征。
27.图1为实施例4所得b、n共同配位的cu单原子催化剂的透射电子显微镜照片,可以看到该样品具有纳米管状表观形貌。
28.图2为实施例4所得b、n共同配位的cu单原子催化剂的扫描透射电子显微镜高角环形暗场像,该视野中并未发现明显聚集型cu颗粒存在,表明实施例4所合成材料中cu原子具有高度分散性,图中白圈标出的亮点即为负载于b、n共同掺杂的c载体上的cu单原子。
29.图3,图4,图5分别为对实施例4所得b、n共同配位的cu单原子催化剂进行软x射线吸收测试所得硼、氮、碳k边吸收谱图。该软x射线吸收测试结果表明所合成的cu单原子催化剂中载体部分为b, n均匀共掺杂的石墨碳结构,其中b和n之间也部分成键。另外,b谱中观察到明显b-o信号,推测材料表面可能存在b-oh物种。
30.图6为对实施例4所得b、n共同配位的cu单原子催化剂中cu元素进行x射线吸收精细结构测试所得谱图,测试结果表明该材料中没有cu-cu信号,即cu原子均是单分散的。进一步对所得cu原子第一配位层结构信号同时进行b和n的配位拟合,拟合结果和原始谱图在第一配未层结构附近完美重合,证实了实施例4所合成材料为b、n共同配位的cu单原子催化剂。
31.实施例5bcn-cu催化剂的co2电化学还原反应催化性能测试采用本发明实施例4所制得的bcn-cu单原子催化剂进行co2电化学还原反应的催化性能测试。
32.co2电化学还原反应的性能测试在如图7所示的流动型电解池中进行,1为负载有实施例4所制得的bcn-cu单原子催化剂的气体扩散电极,2为阳极材料泡沫镍,阴阳两极分别供给0.5m 碳酸氢钾溶液和1m 氢氧化钾溶液作为电解液,3为分隔阴阳两极电解液的质子交换膜,4为用于监测反应中阴极实时电位的参比电极ag/agcl,5为阴极供给反应气co2的进气口,6为出气口。在该流动型电解池中以co2纯气为原料气进行二氧化碳电还原性能测试,测试过程中二氧化碳流速保持为30sccm,阴极碳酸氢钾电解液和阳极氢氧化钾电解液流速分别保持为45ml/h和60ml/h。测试采用恒电流方法,反应气相产物由气相色谱检测,液相产物由核磁共振氢谱检测,计算阴极出气口6的尾气中产物浓度和阴极电解液腔出液口得到液相产物浓度对应的库伦量,根据电化学工作站记录的总库伦量得到催化的选择性、活性等数据。所测不同电流密度下各产物的法拉第效率如图8所示,所测不同电流密度下甲烷法拉第效率和偏电流密度与阴极电位关系如图9所示。
33.实施例1,2和3的催化性能与实施例4无明显差别。
34.应当理解本文所述的例子和实施方式仅为了说明,并不用于限制本发明,本领域技术人员可根据它做出各种修改或变化,凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1