用于评估肿瘤突变负荷的方法和系统与流程
本申请要求2016年2月29日提交的美国临时申请no.62/301,534的权益。上述申请的内容以引用的方式整体并入本文。
发明领域
本发明涉及评估基因改变诸如肿瘤突变负荷的方法。
发明背景
癌细胞在癌症发展和进展期间累积突变。这些突变可以是dna修复、复制或修饰的固有功能失常或暴露于外部诱变剂的结果。某些突变赋予癌细胞生长优势,并且在癌症出现的组织的微环境中被正向选择。虽然优势突变的选择有助于肿瘤发生,但是随着突变发展,产生肿瘤新抗原和随后的免疫识别的可能性也会增加(gubin和schreiber.science350:158-9,2015)。因此,如通过全外显子组测序(wes)所测量的总突变负荷可用于指导患者治疗决策,例如,预测对癌症免疫疗法的持久响应。然而,将基因组研究转变为常规临床实践仍然存在问题,因为全外显子组测序不是广泛可用的并且是昂贵、耗时和技术上具有挑战性的。
因此,仍然存在对新型方法的需要,所述方法包括靶向基因组或外显子组子集的基因组谱分析(genomicprofiling),以精确地测量肿瘤样品中的突变负荷。
技术实现要素:
本发明至少部分地基于以下发现:例如使用基于杂交捕获的下一代测序(ngs)平台对来自患者样品的一小部分基因组或外显子组谱分析,起到有效替代总突变负荷分析的作用。与例如全基因组或全外显子组测序相比,使用包括靶向ngs方法来检测突变负荷的方法具有若干个优点,包括但不限于更快,例如更易临床管理的周转时间(大约2周),标准化信息学流程以及更易管理的成本。本文公开的方法具有优于传统标记物诸如通过组织化学检测到的蛋白质表达的其他优点,因为本方法产生客观量度(例如,突变负荷)而不是主观量度(例如,病理评分)。本文公开的方法还允许同时检测靶向疗法的可操作改变(actionablealteration),以及免疫疗法的突变负荷。这些方法可为癌症患者的疗法响应提供临床可操作的预测因子。
因此,本发明至少部分地提供了评估样品中的突变负荷的方法,所述方法通过以下方式来实现:从样品中提供亚基因组间隔集合的序列;并且确定突变负荷的值,其中值是亚基因组间隔集合中改变的数量的函数。在某些实施方案中,亚基因组间隔集合来自预定的基因集合,例如,不包括整个基因组或外显子组的预定的基因集合。在某些实施方案中,亚基因组间隔集合是编码亚基因组间隔集合。在其他实施方案中,亚基因组间隔集合包含编码亚基因组间隔和非编码亚基因组间隔。在某些实施方案中,突变负荷的值是亚基因组间隔集合中改变(例如,体细胞改变)的数量的函数。在某些实施方案中,改变的数量排除功能改变、种系改变或两者。在一些实施方案中,样品是肿瘤样品或源自肿瘤的样品。本文描述的方法还可包括例如以下的一种或多种:从样品中获取包括多个肿瘤成员的文库;通过杂交使文库与诱饵集合接触以提供选择的肿瘤成员,从而提供文库捕获;从文库捕获中获取来自肿瘤成员的包含改变的亚基因组间隔的读段;通过比对方法来比对读段;将来自读段的核苷酸值分配给预选的核苷酸位置;并且从分配的核苷酸位置集合中选择亚基因组间隔集合,其中亚基因组间隔集合来自预定的基因集合。
在一个方面,本发明的特征在于一种评估样品,例如肿瘤样品(例如,从肿瘤中获取的样品)中的突变负荷的方法。该方法包括:
a)从样品中提供亚基因组间隔(例如,编码亚基因组间隔)集合的序列,例如核苷酸序列,其中亚基因组间隔集合来自预定的基因集合;以及
b)确定突变负荷的值,其中值是亚基因组间隔集合中改变(例如,一个或多个改变),例如体细胞改变(例如,一个或多个体细胞改变)的数量的函数。
在某些实施方案中,改变的数量排除亚基因组间隔中的功能改变。在其他实施方案中,改变的数量排除亚基因组间隔中的种系改变。在某些实施方案中,改变的数量排除亚基因组间隔中的功能改变和亚基因组间隔中的种系改变。
在某些实施方案中,亚基因组间隔集合包含编码亚基因组间隔。在其他实施方案中,亚基因组间隔集合包含非编码亚基因组间隔。在某些实施方案中,亚基因组间隔集合包含编码亚基因组间隔。在其他实施方案中,亚基因组间隔集合包含一个或多个编码亚基因组间隔和一个或多个非编码亚基因组间隔。在某些实施方案中,亚基因组间隔集合中约5%或更多、约10%或更多、约20%或更多、约30%或更多、约40%或更多、约50%或更多、约60%或更多、约70%或更多、约80%或更多、约90%或更多、或约95%或更多的亚基因组间隔是编码亚基因组间隔。在其他实施方案中,亚基因组间隔集合中约90%或更少、约80%或更少、约70%或更少、约60%或更少、约50%或更少、约40%或更少、约30%或更少、约20%或更少、约10%或更少、或约5%或更少的亚基因组间隔是非编码亚基因组间隔。
在其他实施方案中,亚基因组间隔集合不包括整个基因组或整个外显子组。在其他实施方案中,编码亚基因组间隔集合不包括整个外显子组。
在某些实施方案中,预定的基因集合不包括整个基因组或整个外显子组。在其他实施方案中,预定的基因集合包括表1-4或图3a-4d中所示的一种或多种基因或由所述一种或多种基因组成。
在某些实施方案中,值表示为预定的基因集合的函数。在某些实施方案中,值表示为预定的基因集合的编码区的函数。在其他实施方案中,值表示为预定的基因集合的非编码区的函数。在某些实施方案中,值表示为预定的基因集合的外显子的函数。在其他实施方案中,值表示为预定的基因集合的内含子的函数。
在某些实施方案中,值表示为测序的预定基因集合的函数。在某些实施方案中,值表示为测序的预定基因集合的编码区的函数。在其他实施方案中,值表示为测序的预定基因集合的非编码区的函数。在某些实施方案中,值表示为测序的预定基因集合的外显子的函数。在其他实施方案中,值表示为测序的预定基因集合的内含子的函数。
在某些实施方案中,值表示为预定的基因集合的预选数量位置中的改变(例如,体细胞改变)的数量的函数。在某些实施方案中,值表示为预定的基因集合的编码区的预选数量位置中的改变(例如,体细胞改变)的数量的函数。在其他实施方案中,值表示为预定的基因集合的非编码区的预选数量位置中的改变(例如,体细胞改变)的数量的函数。在某些实施方案中,值表示为预定的基因集合的外显子的预选数量位置中的改变(例如,体细胞改变)的数量的函数。在其他实施方案中,值表示为预定的基因集合的内含子的预选数量位置中的改变(例如,体细胞改变)的数量的函数。
在某些实施方案中,值表示为测序的预定基因集合的预选数量位置中的改变(例如,体细胞改变)的数量的函数。在某些实施方案中,值表示为测序的预定基因集合的编码区的预选数量位置中的改变(例如,体细胞改变)的数量的函数。在其他实施方案中,值表示为测序的预定基因集合的非编码区的预选数量位置中的改变(例如,体细胞改变)的数量的函数。在某些实施方案中,值表示为测序的预定基因集合的外显子的预选数量位置中的改变(例如,体细胞改变)的数量的函数。在其他实施方案中,值表示为测序的预定基因集合的内含子的预选数量位置中的改变(例如,体细胞改变)的数量的函数。
在某些实施方案中,值表示为每个预选单位的改变(例如,体细胞改变)的数量的函数,例如,表示为每兆碱基的体细胞改变的数量的函数。
在某些实施方案中,值表示为预定的基因集合中每兆碱基的改变(例如,体细胞改变)的数量的函数。在某些实施方案中,值表示为预定的基因集合的编码区中每兆碱基的改变(例如,体细胞改变)的数量的函数。在其他实施方案中,值表示为预定的基因集合的非编码区中每兆碱基的改变(例如,体细胞改变)的数量的函数。在某些实施方案中,值表示为预定的基因集合的外显子中每兆碱基的改变(例如,体细胞改变)的数量的函数。在其他实施方案中,值表示为预定的基因集合的内含子中每兆碱基的改变(例如,体细胞改变)的数量的函数。
在某些实施方案中,值表示为测序的预定基因集合中每兆碱基的改变(例如,体细胞改变)的数量的函数。在某些实施方案中,值表示为测序的预定基因集合的编码区中每兆碱基的改变(例如,体细胞改变)的数量的函数。在其他实施方案中,值表示为测序的预定基因集合的非编码区中每兆碱基的改变(例如,体细胞改变)的数量的函数。在某些实施方案中,值表示为测序的预定基因集合的外显子中每兆碱基的改变(例如,体细胞改变)的数量的函数。在其他实施方案中,值表示为测序的预定基因集合的内含子中每兆碱基的改变(例如,体细胞改变)的数量的函数。
在某些实施方案中,将突变负荷外推至较大部分的基因组,例如外推至外显子组或整个基因组,例如以便获得总突变负荷。在其他实施方案中,将突变负荷外推至较大部分的外显子组,例如,外推至整个外显子组。
在某些实施方案中,样品来自受试者。在某些实施方案中,受试者患有病症,例如癌症。在其他实施方案中,受试者正在接受或已接受疗法,例如免疫疗法。
在某些实施方案中,突变负荷表示为例如来自参考群体的样品中的突变负荷之中的百分位数。在某些实施方案中,参考群体包括与受试者患有相同类型癌症的患者。在其他实施方案中,参考群体包括正在接受或已接受与受试者相同类型疗法的患者。
另一方面,本发明的特征在于一种评估样品,例如肿瘤样品或源自肿瘤的样品中的突变负荷的方法。方法包括:
(i)从样品中获取包括多个肿瘤成员的文库;
(ii)使文库与诱饵集合接触以提供选择的肿瘤成员,其中所述诱饵集合与肿瘤成员杂交,从而提供文库捕获;
(iii)例如通过下一代测序方法,从所述文库捕获中获取来自肿瘤成员的包含改变(例如,体细胞改变)的亚基因组间隔的读段;
(iv)通过比对方法来比对所述读段;
(v)将来自所述读段的核苷酸值分配给预选的核苷酸位置;
(vi)从分配的核苷酸位置集合中选择亚基因组间隔(例如,编码亚基因组间隔)集合,其中亚基因组间隔集合来自预定的基因集合;以及
(vii)确定突变负荷的值,其中值是亚基因组间隔集合中改变(例如,一个或多个改变),例如体细胞改变(例如,一个或多个体细胞改变)的数量的函数。
在某些实施方案中,改变(例如,体细胞改变)的数量排除亚基因组间隔中的功能改变。在其他实施方案中,改变的数量排除亚基因组间隔中的种系改变。在某些实施方案中,改变(例如,体细胞改变)的数量排除亚基因组间隔中的功能改变和亚基因组间隔中的种系改变。
改变类型
在本文所述的方法或系统中,可评估各种类型的改变(例如,体细胞改变)并且用于突变负荷的分析。
体细胞改变
在某些实施方案中,根据本文描述的方法评估的改变是一种改变(例如,体细胞改变)。
在某些实施方案中,改变(例如,体细胞改变)是编码短变体,例如碱基取代或插入缺失(插入或缺失)。在某些实施方案中,改变(例如,体细胞改变)是点突变。在其他实施方案中,改变(例如,体细胞改变)不是重排,例如不是易位。在某些实施方案中,改变(例如,体细胞改变)是剪接变体。
在某些实施方案中,改变(例如,体细胞改变)是沉默突变,例如同义改变。在其他实施方案中,改变(例如,体细胞改变)是非同义单核苷酸变体(snv)。在其他实施方案中,改变(例如,体细胞改变)是乘客突变,例如对细胞克隆的适应性具有不可检测影响的改变。在某些实施方案中,改变(例如,体细胞改变)是意义不明变体(vus),例如其致病性既不能被确认也不能被排除的改变。在某些实施方案中,改变(例如,体细胞改变)尚未被鉴定为与癌症表型相关。
在某些实施方案中,改变(例如,体细胞改变)与对细胞分裂、生长或存活的影响无关或不知道与其相关。在其他实施方案中,改变(例如,体细胞改变)与对细胞分裂、生长或存活的影响相关。
在某些实施方案中,增加的体细胞改变水平是增加的一种或多种类别或类型体细胞改变(例如,重排、点突变、插入缺失或其任何组合)水平。在某些实施方案中,增加的体细胞改变水平是增加的一种类别或类型体细胞改变(例如,仅重排、仅点突变或仅插入缺失)水平。在某些实施方案中,增加的体细胞改变水平是增加的预选位置处的体细胞改变(例如,本文描述的改变)水平。在某些实施方案中,增加的体细胞改变水平是增加的预选体细胞改变(例如,本文描述的改变)水平。
功能改变
在某些实施方案中,改变(例如,体细胞改变)的数量排除亚基因组间隔中的功能改变。
在一些实施方案中,功能改变是与参考序列例如野生型或未突变的序列相比,对细胞分裂、生长或存活具有影响例如促进细胞分裂、生长或存活的改变。在某些实施方案中,功能改变本身通过包括在功能改变的数据库例如cosmic数据库(cancer.sanger.ac.uk/cosmic;forbes等nucl.acidsres.2015;43(d1):d805-d811)中来鉴定。在其他实施方案中,功能改变是具有已知功能状态,例如作为cosmic数据库中已知体细胞改变而存在的改变。在某些实施方案中,功能改变是具有可能的功能状态,例如肿瘤抑制基因中的截短的改变。在某些实施方案中,功能改变是司机突变,例如,例如通过增加细胞存活或繁殖而为其微环境中的克隆提供选择性优势的改变。在其他实施方案中,功能改变是能够引起克隆扩增的改变。在某些实施方案中,功能改变是能够引起以下的一种、两种、三种、四种、五种或所有的改变:(a)生长信号自给自足;(b)抗生长信号减少,例如对抗生长信号不灵敏;(c)细胞凋亡减少;(d)复制潜力增加;(e)持续的血管生成;或(f)组织浸润或转移。
在某些实施方案中,功能改变不是乘客突变,例如不是对细胞克隆的适应性具有不可检测影响的改变。在某些实施方案中,功能改变不是意义不明变体(vus),例如不是其致病性既不能被确认也不能被排除的改变。
在某些实施方案中,排除预定的基因集合中的预选肿瘤基因中的多个(例如,约10%、20%、30%、40%、50%、60%、70%、80%、90%或更多)功能改变。在某些实施方案中,排除预定的基因集合中的预选基因(例如,肿瘤基因)中的所有功能改变。在某些实施方案中,排除预定的基因集合中的多个预选基因(例如,肿瘤基因)中的多个功能改变。在某些实施方案中,排除预定的基因集合中的所有基因(例如,肿瘤基因)中的所有功能改变。
种系突变
在某些实施方案中,改变的数量排除亚基因组间隔中的种系突变。在某些实施方案中,体细胞改变不同于或类似于,例如区别于种系突变。
在某些实施方案中,种系改变是单核苷酸多态性(snp)、碱基取代、插入缺失(例如,插入或缺失)或沉默突变(例如,同义突变)。
在某些实施方案中,通过使用一种方法来排除种系改变,所述方法不使用与匹配的正常序列比较。在其他实施方案中,通过包括使用sgz算法的方法来排除种系改变。在某些实施方案中,种系改变本身通过包括在种系改变的数据库例如dbsnp数据库(www.ncbi.nlm.nih.gov/snp/index.html;sherry等nucleicacidsres.2001;29(1):308-311)中来鉴定。在其他实施方案中,种系改变本身通过包括在exac数据库的两项或更多项(exac.broadinstitute.org;exomeaggregationconsortium等“analysisofprotein-codinggeneticvariationin60,706humans,”biorxivpreprint.2015年10月30日)中来鉴定。在一些实施方案中,种系改变本身通过包括在千人基因组计划数据库(www.1000genomes.org;mcvean等nature.2012;491,56–65)中来鉴定。在一些实施方案中,种系改变本身通过包括在esp数据库(外显子组变异数据库,nhlbigo外显子组测序计划(esp),seattle,wa(evs.gs.washington.edu/evs/)中来鉴定。
多基因分析
本文描述的方法和系统评估例如亚基因组间隔集合,所述亚基因组间隔集合例如来自预定的基因集合。
在某些实施方案中,预定的基因集合包括多个基因,所述多个基因以突变形式与对细胞分裂、生长或存活的影响相关,或与癌症例如本文描述的癌症相关。
在某些实施方案中,预定的基因集合包括例如本文所述的至少约50或更多、约100或更多、约150或更多、约200或更多、约250或更多、约300或更多、约350或更多、约400或更多、约450或更多、约500或更多、约550或更多、约600或更多、约650或更多、约700或更多、约750或更多、或约800或更多个基因。在一些实施方案中,预定的基因集合包括选自表1-4或图3a-4d的至少约50或更多、约100或更多、约150或更多、约200或更多、约250或更多、约300或更多个、或所有的基因或基因产物。
在某些实施方案中,方法还包括从样品中获取包括多个肿瘤成员的文库。在某些实施方案中,方法还包括使文库与诱饵集合接触以提供选择的肿瘤成员,其中所述诱饵集合与来自文库的肿瘤成员杂交,从而提供文库捕获。在某些实施方案中,方法还包括例如通过下一代测序方法,从文库或文库捕获中获取来自肿瘤成员的包含改变(例如,体细胞改变)的亚基因组间隔的读段,从而获取亚基因组间隔的读段。在某些实施方案中,方法还包括通过比对方法,例如本文描述的比对方法来比对亚基因组间隔的读段。在某些实施方案中,方法还包括例如通过本文描述的突变识别方法,从亚基因组间隔的读段中将核苷酸值分配给预选的核苷酸位置。
在某些实施方案中,方法还包括以下的一种、两种、三种、四种或所有:
(a)从样品中获取包括多个肿瘤成员的文库;
(b)使文库与诱饵集合接触以提供选择的肿瘤成员,其中所述诱饵集合与肿瘤成员杂交,从而提供文库捕获;
(c)例如通过下一代测序方法,从所述文库捕获中获取来自肿瘤成员的包含改变(例如,体细胞改变)的亚基因组间隔的读段,从而获取亚基因组间隔的读段;
(d)通过比对方法,例如本文描述的比对方法来比对所述读段;或
(e)例如通过本文描述的突变识别方法,将来自所述读段的核苷酸值分配给预选的核苷酸位置。
在某些实施方案中,获取亚基因组间隔的读段包括对亚基因组间隔进行测序,所述亚基因组间隔来自至少约50或更多、约100或更多、约150或更多、约200或更多、约250或更多、约300或更多、约350或更多、约400或更多、约450或更多、约500或更多、约550或更多、约600或更多、约650或更多、约700或更多、约750或更多、或约800或更多个基因。在某些实施方案中,获取亚基因组间隔的读段包括对亚基因组间隔进行测序,所述亚基因组间隔来自选自表1-4或图3a-4d的至少约50或更多、约100或更多、约150或更多、约200或更多、约250或更多、约300或更多个、或所有的基因或基因产物。
在某些实施方案中,获取亚基因组间隔的读段包括用大于约250x的平均独特覆盖度进行测序。在其他实施方案中,获取亚基因组间隔的读段包括用大于约500x的平均独特覆盖度进行测序。在其他实施方案中,获取亚基因组间隔的读段包括用大于约1,000x的平均独特覆盖度进行测序。
在某些实施方案中,获取亚基因组间隔的读段包括在测序的大于约99%的基因(例如,外显子)处用大于约250x的平均独特覆盖度进行测序。在其他实施方案中,获取亚基因组间隔的读段包括在测序的大于约95%的基因(例如,外显子)处用大于约500x的平均独特覆盖度进行测序。在某些实施方案中,获取亚基因组间隔的读段包括在测序的大于约99%的基因(例如,外显子)处用大于约250x、大于约500x、或大于约1,000x的平均独特覆盖度进行测序。
在某些实施方案中,通过本文描述的方法提供本文描述的亚基因组间隔(例如,编码亚基因组间隔)集合的序列,例如核苷酸序列。在某些实施方案中,在不需要使用包括匹配的正常对照(例如,野生型对照)、匹配的肿瘤对照(例如,原发性与转移性)或两者的方法的情况下提供序列。
sgz分析
在某些实施方案中,通过包括使用sgz算法的方法或系统来排除种系改变。
在某些实施方案中,方法还包括通过以下表征肿瘤样品中的变体,例如改变:
a)获取:
i)序列覆盖度输入(sci),其对于多个选择的亚基因组间隔中的每一个包括在选择的亚基因组间隔处归一化的序列覆盖度的值,其中sci是亚基因组间隔的读段数量和方法匹配的对照的读段数量的函数;
ii)snp等位基因频率输入(safi),其对于多个选择的种系snp中的每一个包括肿瘤样品中等位基因频率的值,其中safi至少部分地基于肿瘤样品中次要或替代等位基因频率;以及
iii)变体等位基因频率输入(vafi),其包括肿瘤样品中所述变体的等位基因频率;
b)获取作为sci和safi的函数的以下值:
i)多个基因组区段中的每一个的基因组区段总拷贝数(c);
ii)多个基因组区段中的每一个的基因组区段次要等位基因拷贝数(m);以及
iii)样品纯度(p),
其中c、m和p的值通过将全基因组拷贝数模型拟合至sci和safi来获得;以及
c)获取:
突变类型g的值,其表示变体是体细胞的、亚克隆体细胞变体、种系的或不可辨识的,并且是vafi、p、c和m的函数。
在某些实施方案中,方法还包括对多个选择的亚基因组间隔中的每一个、多个选择的种系snp中的每一个和变体(例如,改变)进行测序,其中在归一化之前的平均序列覆盖度为至少约250x,例如至少约500x。
在某些实施方案中,将全基因组拷贝数模型拟合至sci包括使用以下方程:
在某些实施方案中,将全基因组拷贝数模型拟合至safi包括使用以下方程:
在某些实施方案中,通过确定vafi、p、c和m的值与体细胞/种系状态的模型的拟合来确定g。在某些实施方案中,通过以下来获取g值:
在某些实施方案中,为0或接近0的g值表示变体是体细胞变体;为1或接近1的g值表示变体是种系变体;小于1但大于0的g值表示不可辨识的结果;或显著小于0的g值表示变体是亚克隆体细胞变体。
sgz算法描述于国际申请公布wo2014/183078和美国申请公布2014/0336996中,所述申请公布的内容以引用的方式整体并入。sgz算法也描述于sun等cancerresearch2014;74(19s):1893-1893中。
样品,例如肿瘤样品
本文描述的方法和系统可用于评估来自许多不同来源的各种类型样品中的突变负荷。
在一些实施方案中,样品是肿瘤样品或源自肿瘤的样品。在某些实施方案中,样品从实体瘤、血液学癌症或其转移形式中获取。在某些实施方案中,如本文所述,样品从患有癌症的受试者、或正在接受疗法或已接受疗法的受试者中获得。
在一些实施方案中,样品(例如,肿瘤样品)包含以下的一种或多种:恶化前或恶性细胞;来自实体瘤、软组织肿瘤或转移病变的细胞;来自手术切缘的组织或细胞;组织学正常的组织;一个或多个循环肿瘤细胞(ctc);正常相邻组织(nat);来自患有肿瘤或处于患有肿瘤风险的相同受试者的血液样品;或ffpe样品。在某些实施方案中,样品包含循环肿瘤dna(ctdna)。
在某些实施方案中,样品是ffpe样品。在某些实施方案中,ffpe样品具有以下特性中的一种、两种或所有:(a)表面积为约10mm2或更大、约25mm2或更大、或约50mm2或更大;(b)样品体积为约1mm3或更大、约2mm3或更大、约3mm3或更大、约4mm3或更大、或约5mm3或更大;或(c)有核细胞结构为约50%或更多、约60%或更多、约70%或更多、约80%或更多、或约90%或更多、或约10,000个细胞或更多、约20,000个细胞或更多、约30,000个细胞或更多、约40,000个细胞或更多、或约50,000个细胞或更多。
系统
另一方面,本发明的特征在于一种用于评估样品(例如,肿瘤样品或源自肿瘤的样品)中的突变负荷的系统。系统包括可操作地连接至存储器的至少一个处理器,当执行时,至少一个处理器被配置为:
a)从样品中获取亚基因组间隔(例如,编码亚基因组间隔)集合的序列,例如核苷酸序列,其中编码亚基因组间隔集合来自预定的基因集合;以及
b)确定突变负荷的值,其中值是亚基因组间隔集合中改变(例如,体细胞改变)的数量的函数。
在某些实施方案中,所述改变的数量排除:(i)亚基因组间隔(例如,编码亚基因组间隔)中的功能改变,(ii)亚基因组间隔(例如,编码亚基因组间隔)中的种系改变,或(iii)两者。
应用
在一些实施方案中,方法还包括响应于突变负荷的评估,例如增加的突变负荷水平选择治疗。在一些实施方案中,方法还包括响应于突变负荷的评估,例如增加的突变负荷水平施用治疗。在一些实施方案中,方法还包括响应于突变负荷的评估,对样品或样品源自其中的受试者进行分类。在一些实施方案中,方法还包括向患者或向另一个人或主体、护理人员、医生、肿瘤学家、医院、诊所、第三方付款人、保险公司或政府办公室生成和传递报告,例如电子的、基于网络的或纸质的报告。在一些实施方案中,报告包括来自方法的输出,所述输出包括突变负荷。
本发明的另外方面或实施方案包括以下内容的一种或多种。
比对
本文公开的方法可合并使用多种单独调整的比对方法或算法来优化测序方法中的性能,特别是在依赖于大量不同基因中的大量不同遗传事件的大规模平行测序的方法中,例如分析例如来自本文描述的癌症的肿瘤样品的方法。在实施方案中,使用多种比对方法来分析读段,所述多种比对方法针对不同基因中的许多变体中的每一个单独地定制或调整。在实施方案中,调整可以是以下的(一种或多种的)函数:被测序的基因(或其他亚基因组间隔)、样品中的肿瘤类型、被测序的变体、或样品或受试者的特征。选择或使用针对待测序的许多受试者间隔(例如,亚基因组间隔、表达的亚基因组间隔、或两者)单独调整的比对条件允许优化速度、灵敏度和特异性。当优化相对大量的不同受试者间隔(例如,亚基因组间隔、表达的亚基因组间隔、或两者)的读段比对时,方法特别有效。
因此,在一个方面,本发明的特征在于一种分析样品的方法,所述样品例如来自恶性(或恶化前)血液病的肿瘤样品,例如本文描述的恶性(或恶化前)血液病。方法包括:
(a)获取一个或多个文库,所述一个或多个文库包括来自样品的多个成员,例如来自肿瘤样品的多个肿瘤成员;
(b)任选地,例如通过使一个或多个文库与诱饵集合(或多个诱饵集合)接触以提供选择的成员(在本文中有时称为文库捕获),使一个或多个文库富含预选序列;
(c)例如通过包括测序的方法,例如使用下一代测序方法,从文库或文库捕获中获取来自成员例如肿瘤成员的受试者间隔例如亚基因组间隔或表达的亚基因组间隔的读段;
(d)通过比对方法,例如本文描述的比对方法来比对所述读段;以及
(e)将来自所述读段的核苷酸值分配(例如识别突变,例如使用贝叶斯方法)给预选的核苷酸位置,
从而分析所述肿瘤样品,
任选地,其中:
使用独特比对方法来比对来自x个独特受试者间隔(例如,亚基因组间隔、表达的亚基因组间隔、或两者)中的每一个的读段,其中独特受试者间隔(例如,亚基因组间隔或表达的亚基因组间隔)意味着不同于其他x-1个受试者间隔(例如,亚基因组间隔、表达的亚基因组间隔、或两者),并且其中独特比对方法意味着不同于其他x-1种比对方法,并且x为至少2。
在一个实施方案中,方法包括获取文库,从所述文库中各自获得对应于亚基因组间隔的成员和对应于表达的亚基因组间隔的成员。
在一个实施方案中,方法包括获取第一文库,从所述第一文库中获得对应于亚基因组间隔的成员,并且获取第二文库,从所述第二文库中获得对应于表达的亚基因组间隔的成员。
在一个实施方案中,诱饵集合用于提供包含亚基因组间隔和表达间隔的成员或文库捕获。
在一个实施方案中,第一诱饵集合用于提供包含亚基因组间隔的成员或文库捕获,并且第二诱饵集合用于提供包含表达的亚基因组间隔的成员或文库捕获。
在一个实施方案中,存在步骤(b)。在一个实施方案中,不存在步骤(b)。
在一个实施方案中,x为至少3、4、5、10、15、20、30、50、100、200、300、400、500、600、700、800、900或1,000。
在一个实施方案中,使用独特比对方法来比对来自至少x个基因,例如来自表1-4或图3a-4d的至少x个基因的受试者间隔(例如,亚基因组间隔、表达的亚基因组间隔、或两者),并且x等于2、3、4、5、10、15、20、30、40、50、60、70、80、90、100、200、300、400、500或更大。
在一个实施方案中,方法(例如,以上叙述的方法的要素(d))包括选择或使用用于分析例如比对读段的比对方法,
其中所述比对方法是以下的一种或多种或所有的函数,响应于以下的一种或多种或所有而选择,或针对以下的一种或多种或所有而优化:
(i)肿瘤类型,例如所述样品中的肿瘤类型;
(ii)基因或基因类型,被测序的所述受试者间隔(例如,亚基因组间隔或表达的亚基因组间隔)位于其中,例如通过预选变体或变体类型例如突变或通过具有预选频率的突变来表征的基因或基因类型;
(iii)被分析的位点(例如,核苷酸位置);
(iv)被评估的受试者间隔(例如,亚基因组间隔或表达的亚基因组间隔)内的变体类型,例如取代;
(v)样品类型,例如ffpe样品、血液样品或骨髓抽吸物样品;以及
(vi)被评估的所述亚基因组间隔中或附近的序列,例如所述受试者间隔(例如,亚基因组间隔或表达的亚基因组间隔)的预期错位倾向,例如在所述受试者间隔(例如,亚基因组间隔或表达的亚基因组间隔)中或附近存在重复序列。
如本文其他地方所提及,当优化相对大量的受试者间隔(例如,亚基因组间隔、表达的亚基因组间隔、或两者)的读段比对时,方法特别有效。因此,在一个实施方案中,至少x种独特比对方法用于分析至少x个独特亚基因组间隔的读段,其中独特意味着不同于另外的x-1,并且x等于2、3、4、5、10、15、20、30、50、100、200、300、400、500、600、700、800、900、1,000或更大。
在一个实施方案中,分析来自表1-4或图3a-4d的至少x个基因的受试者间隔(例如,亚基因组间隔、表达的亚基因组间隔、或两者),并且x等于2、3、4、5、10、15、20、30、40、50、60、70、80、90、100、200、300、400、500或更大。
在一个实施方案中,将独特比对方法应用于至少3、5、10、20、40、50、60、70、80、90、100、200、300、400或500个不同基因中的每一个中的受试者间隔(例如,亚基因组间隔、表达的亚基因组间隔、或两者)。
在一个实施方案中,给至少20、40、60、80、100、120、140、160或180、200、300、400、或500个基因例如来自表1-4或图3a-4d的基因中的核苷酸位置分配核苷酸值。在一个实施方案中,将独特比对方法应用于分析的所述基因的至少10%、20%、30%、40%、或50%中的每一个中的受试者间隔(例如,亚基因组间隔或表达的亚基因组间隔)。
本文公开的方法允许快速且有效比对麻烦的读段,例如具有重排的读段。因此,在其中受试者间隔(例如,亚基因组间隔或表达的亚基因组间隔)的读段包括具有重排例如易位的核苷酸位置的一个实施方案中,方法可包括使用一种比对方法,所述比对方法被适当地调整并且包括:
选择用于与读段比对的重排参考序列,其中预选所述重排参考序列来与预选重排(在实施方案中,参考序列与基因组重排不同)比对;
将读段与所述预选重排参考序列进行比较,例如比对。
在实施方案中,使用其他方法来比对麻烦的读段。当优化相对大量的不同亚基因组间隔的读段比对时,这些方法特别有效。举例来说,分析肿瘤样品的方法可包括:
在第一组参数(例如,第一作图算法或使用第一参考序列)下执行读段的比较例如比对比较,并且确定所述读段是否符合第一预定比对标准(例如,可将读段与所述第一参考序列比对,所述第一参考序列例如具有小于预选数量的错配);
如果所述读段不符合第一预定比对标准,则在第二组参数(例如,第二作图算法或使用第二参考序列)下执行第二比对比较;并且
任选地,确定所述读段是否符合所述第二预定标准(例如,可将读段与具有小于预选数量的错配的所述第二参考序列比对),
其中所述第二组参数包括使用例如所述第二参考序列的一组参数,与所述第一组参数相比,所述第二组参数更可能产生与预选变体,例如重排、例如插入、缺失或易位的读段比对。
这些和其他比对方法在本文其他地方更详细地讨论,例如在具体实施方式中的标题为“比对”的章节中。所述模块的要素可包括在分析肿瘤的方法中。在实施方案中,将来自标题为“比对”的章节(在发明内容和/或具体实施方式中)的比对方法与来自标题为“突变识别”的章节(在发明内容和/或具体实施方式中)的突变识别方法和/或来自标题为“诱饵”的章节(在发明内容中)和/或标题为“诱饵的设计和构建”和“诱饵合成”的章节(在具体实施方式中)的诱饵集合组合。方法可应用于来自标题为“基因选择”的章节(在发明内容和/或具体实施方式中)的受试者间隔(例如,亚基因组间隔、表达的亚基因组间隔、或两者)集合。
突变识别
本文公开的方法可合并使用定制或调整的突变识别参数来优化测序方法中的性能,特别是在依赖于大量不同基因中的大量不同遗传事件的大规模平行测序的方法中,所述基因例如来自肿瘤样品(例如,来自本文描述的癌症)。在方法的实施方案中,许多预选受试者间隔(例如,亚基因组间隔、表达的亚基因组间隔、或两者)中的每一个的突变识别都被单独定制或微调。定制或调整可基于本文描述的一个或多个因素,例如样品中的癌症类型、待测序的受试者间隔(例如,亚基因组间隔或表达的亚基因组间隔)位于其中的基因、或待测序的变体。这种选择或使用针对待测序的许多受试者间隔(例如,亚基因组间隔、表达的亚基因组间隔、或两者)精调的比对条件允许优化速度、灵敏度和特异性。当优化相对大量的不同受试者间隔(例如,亚基因组间隔、表达的亚基因组间隔、或两者)的读段比对时,方法特别有效。
因此,在一个方面,本发明的特征在于一种分析样品的方法,所述样品例如来自恶性(或恶化前)血液病的肿瘤样品,例如本文描述的恶性(或恶化前)血液病。方法包括:
(a)获取一个或多个文库,所述一个或多个文库包括来自样品的多个成员,例如来自样品(例如肿瘤样品)的多个肿瘤成员;
(b)任选地,例如通过使文库与诱饵集合(或多个诱饵集合)接触以提供选择的成员例如文库捕获,使一个或多个文库富含预选序列;
(c)例如通过包括测序的方法,例如使用下一代测序方法,从所述文库或文库捕获中获取来自成员例如肿瘤成员的受试者间隔(例如,亚基因组间隔或表达的亚基因组间隔)的读段;
(d)通过比对方法,例如本文描述的比对方法来比对所述读段;以及
(e)将来自所述读段的核苷酸值分配(例如识别突变,例如使用贝叶斯方法或本文描述的识别方法)给预选的核苷酸位置,从而分析所述肿瘤样品。
任选地,其中将核苷酸值分配给由独特识别方法分配的x个独特受试者间隔(亚基因组间隔、表达的亚基因组间隔、或两者)中的每一个中的核苷酸位置,其中独特受试者间隔(例如,亚基因组间隔或表达的亚基因组间隔)意味着不同于其他x-1个受试者间隔(例如,亚基因组间隔、表达的亚基因组间隔、或两者),并且其中独特识别方法意味着不同于其他x-1种识别方法,并且x为至少2。识别方法可以不同,因此例如通过依赖于不同的贝叶斯先验值(priorvalue)是独特的。
在一个实施方案中,方法包括获取文库,从所述文库中各自获得对应于亚基因组间隔的成员和对应于表达的亚基因组间隔的成员。
在一个实施方案中,方法包括获取第一文库,从所述第一文库中获得对应于亚基因组间隔的成员,并且获取第二文库,从所述第二文库中获得对应于表达的亚基因组间隔的成员。
在一个实施方案中,诱饵集合用于提供包含亚基因组间隔和表达间隔的成员或文库捕获。
在一个实施方案中,第一诱饵集合用于提供包含亚基因组间隔的成员或文库捕获,并且第二诱饵集合用于提供包含表达的亚基因组间隔的成员或文库捕获。
在一个实施方案中,存在步骤(b)。在一个实施方案中,不存在步骤(b)。
在一个实施方案中,分配所述核苷酸值是一种值的函数,所述值作为或代表观察到读段在一种类型的肿瘤中的所述预选核苷酸位置处示出预选变体例如突变的先验(例如,文献)期望值。
在一个实施方案中,方法包括将核苷酸值分配(例如,识别突变)给至少10、20、40、50、60、70、80、90、100、200、300、400、500、600、700、800、900或1,000个预选核苷酸位置,其中每个分配都是独特(与其他分配完全不同)的值的函数,所述独特的值作为或代表观察到读段在一种类型的肿瘤中的所述预选核苷酸位置处示出预选变体例如突变的先验(例如,文献)期望值。
在一个实施方案中,分配所述核苷酸值是一组值的函数,所述一组值代表如果变体以一定频率(例如,1%、5%、10%等)存在于样品中和/或如果不存在变体(例如,单独由于碱基识别错误在读段中观察到的),观察到读段在所述预选核苷酸位置处示出所述预选变体的概率。
在一个实施方案中,方法(例如,以上叙述的方法的步骤(e))包括突变识别方法。本文描述的突变识别方法可包括以下:
针对所述x个受试者间隔(例如,亚基因组间隔、表达的亚基因组间隔、或两者)中的每一个中的预选核苷酸位置,获取:
(i)第一值,其作为或代表观察到读段在类型x的肿瘤中的所述预选核苷酸位置处示出预选变体例如突变的先验(例如,文献)期望值;以及
(ii)第二组值,其代表如果变体以一定频率(例如,1%、5%、10%等)存在于样品中和/或如果不存在变体(例如,单独由于碱基识别错误在读段中观察到的),观察到读段在所述预选核苷酸位置处示出所述预选变体的概率;
响应于所述值,通过加权,例如通过本文描述的贝叶斯方法,将来自所述读段的核苷酸值分配(例如,识别突变)给所述预选核苷酸位置中的每一个,使用第一值在第二组值之间进行比较(例如,计算突变存在的后验概率),从而分析所述样品。
在一个实施方案中,方法包括以下的一种或多种或所有:
(i)将核苷酸值分配(例如,识别突变)给至少10、20、40、50、60、70、80、90、100、200、300、400、500、600、700、800、900或1,000个预选核苷酸位置,其中每个分配都基于独特(与其他分配完全不同)的第一值和/或第二值;
(ii)(i)的方法的分配,其中使用第一值进行至少10、20、30、40、50、60、70、80、90、100、200、300、400或500个分配,所述第一值是例如预选肿瘤类型中小于5%、10%或20%的细胞存在预选变体的概率的函数;
(iii)将核苷酸值分配(例如,识别突变)给至少x个预选核苷酸位置,所述至少x个预选核苷酸位置中的每一个与预选变体相关,所述预选变体具有独特(与其他x-1个分配完全不同)的存在于预选类型(例如所述样品的肿瘤类型)的肿瘤中的概率,其中,任选地所述x个分配中的每一个都基于独特(与其他x-1个分配完全不同)的第一值和/或第二值(其中x=2、3、5、10、20、40、50、60、70、80、90、100、200、300、400、或500);
(iv)在第一核苷酸位置和第二核苷酸位置处分配核苷酸值(例如,识别突变),其中存在于预选类型(例如,所述样品的肿瘤类型)的肿瘤中的所述第一核苷酸位置处的第一预选变体的概率比存在的所述第二核苷酸位置处的第二预选变体的概率大至少2、5、10、20、30或40倍,其中,任选地每个分配都基于独特(与其他分配完全不同)的第一值和/或第二值;
(v)将核苷酸值分配给多个预选核苷酸位置(例如,识别突变),其中所述多个包括落入以下概率百分比范围中的一个或多个,例如至少3个、4个、5个、6个、7个或所有的对变体的分配:
小于或等于0.01;
大于0.01且小于或等于0.02;
大于0.02且小于或等于0.03;
大于0.03且小于或等于0.04;
大于0.04且小于或等于0.05;
大于0.05且小于或等于0.1;
大于0.1且小于或等于0.2;
大于0.2且小于或等于0.5;
大于0.5且小于或等于1.0;
大于1.0且小于或等于2.0;
大于2.0且小于或等于5.0;
大于5.0且小于或等于10.0;
大于10.0且小于或等于20.0;
大于20.0且小于或等于50.0;以及
大于50且小于或等于100.0%;
其中,概率范围是预选核苷酸位置处的预选变体将存在于预选类型(例如,所述样品的肿瘤类型)的肿瘤中的概率范围,或针对预选类型(例如,所述样品的肿瘤类型),预选核苷酸位置处的预选变体将存在于肿瘤样品中的所述百分比细胞、来自肿瘤样品的文库或来自所述文库的文库捕获中的概率,并且
其中,任选地每个分配都基于独特的第一值和/或第二值(例如,在叙述的概率范围上与其他分配完全不同的独特,或针对其他列出的概率范围中的一个或多个或所有,与第一值和/或第二值完全不同的独特)。
(vi)将核苷酸值分配(例如,识别突变)给至少1、2、3、5、10、20、40、50、60、70、80、90、100、200、300、400、500、600、700、800、900、或1,000个预选核苷酸位置,所述预选核苷酸位置各自独立地具有存在于所述样品中的小于50%、40%、25%、20%、15%、10%、5%、4%、3%、2%、1%、0.5%、0.4%、0.3%、0.2%、或0.1%的dna中的预选变体,其中,任选地每个分配都基于独特(与其他分配完全不同)的第一值和/或第二值;
(vii)在第一核苷酸位置和第二核苷酸位置处分配核苷酸值(例如,识别突变),其中预选变体在所述样品的dna中的第一位置处的概率比预选变体在所述样品的dna中的所述第二核苷酸位置处的概率大至少2、5、10、20、30或40倍,其中,任选地每个分配都基于独特(与其他分配完全不同)的第一值和/或第二值;
(viii)在以下的一种或多种或所有中,分配核苷酸值(例如,识别突变):
(1)来自所述样品的文库中的核酸、或来自所述文库的文库捕获中的核酸的至少1、2、3、4或5个预选核苷酸位置,所述预选核苷酸位置具有存在于所述样品中的小于1%的细胞中的预选变体;
(2)来自所述样品的文库中的核酸、或来自所述文库的文库捕获中的核酸的至少1、2、3、4或5个预选核苷酸位置,所述预选核苷酸位置具有存在于所述样品中的1%-2%的细胞中的预选变体;
(3)来自所述样品的文库中的核酸、或来自所述文库的文库捕获中的核酸的至少1、2、3、4或5个预选核苷酸位置,所述预选核苷酸位置具有存在于所述样品中的大于2%且小于或等于3%的细胞中的预选变体;
(4)来自所述样品的文库中的核酸、或来自所述文库的文库捕获中的核酸的至少1、2、3、4或5个预选核苷酸位置,所述预选核苷酸位置具有存在于所述样品中的大于3%且小于或等于4%的细胞中的预选变体;
(5)来自所述样品的文库中的核酸、或来自所述文库的文库捕获中的核酸的至少1、2、3、4或5个预选核苷酸位置,所述预选核苷酸位置具有存在于所述样品中的大于4%且小于或等于5%的细胞中的预选变体;
(6)来自所述样品的文库中的核酸、或来自所述文库的文库捕获中的核酸的至少1、2、3、4或5个预选核苷酸位置,所述预选核苷酸位置具有存在于所述样品中的大于5%且小于或等于10%的细胞中的预选变体;
(7)来自所述样品的文库中的核酸、或来自所述文库的文库捕获中的核酸的至少1、2、3、4或5个预选核苷酸位置,所述预选核苷酸位置具有存在于所述样品中的大于10%且小于或等于20%的细胞中的预选变体;
(8)来自所述样品的文库中的核酸、或来自所述文库的文库捕获中的核酸的至少1、2、3、4或5个预选核苷酸位置,所述预选核苷酸位置具有存在于所述样品中的大于20%且小于或等于40%的细胞中的预选变体;
(9)来自所述样品的文库中的核酸、或来自所述文库的文库捕获中的核酸的至少1、2、3、4或5个预选核苷酸位置,所述预选核苷酸位置具有存在于所述样品中的大于40%且小于或等于50%的细胞中的预选变体;或
(10)来自所述样品的文库中的核酸、或来自所述文库的文库捕获中的核酸的至少1、2、3、4或5个预选核苷酸位置,所述预选核苷酸位置具有存在于所述样品中的大于50%且小于或等于100%的细胞中的预选变体;
其中,任选地每个分配都基于独特的第一值和/或第二值(例如,在叙述的范围(例如,在(1)中的小于1%的范围)上与其他分配完全不同的独特,或针对确定其他列出的范围中的一个或多个或所有,与第一值和/或第二值完全不同的独特);或
(ix)在x个核苷酸位置中的每一个处分配核苷酸值(例如,识别突变),每个核苷酸位置独立地具有(存在于所述样品的dna中的预选变体的)一种可能性,所述可能性与其他x-1个核苷酸位置处的预选变体的可能性相比是独特的,其中x等于或大于1、2、3、5、10、20、40、50、60、70、80、90、100、200、300、400、500、600、700、800、900、或1,000,并且其中每个分配都基于独特(与其他分配完全不同)的第一值和/或第二值。
在方法的实施方案中,“阈值”用于评估读段,并且从读段中选择核苷酸位置的值,例如在基因中的特定位置处识别突变。在方法的实施方案中,定制或微调针对许多预选受试者间隔(例如,亚基因组间隔、表达的亚基因组间隔、或两者)中的每一个的阈值。定制或调整可基于本文描述的一个或多个因素,例如样品中的癌症类型、待测序的受试者间隔(亚基因组间隔或表达的亚基因组间隔)位于其中的基因、或待测序的变体。这提供了针对待测序的许多受试者间隔(例如,亚基因组间隔、表达的亚基因组间隔、或两者)中的每一个精调的识别。当分析相对大量的不同亚基因组间隔时,方法特别有效。
因此,在另一个实施方案中,分析肿瘤的方法包括以下突变识别方法:
针对所述x个受试者间隔(例如,亚基因组间隔、表达的亚基因组间隔、或两者)中的每一个,获取阈值,其中所述获取的x个阈值中的每一个与其他x-1个阈值相比是独特的,从而提供x个独特阈值;
针对所述x个受试者间隔(亚基因组间隔、表达的亚基因组间隔、或两者)中的每一个,将观察值与其独特的阈值比较,所述观察值是在预选核苷酸位置处具有预选核苷酸值的读段数量的函数,从而将其独特的阈值应用于所述x个受试者间隔(例如,亚基因组间隔、表达的亚基因组间隔、或两者)中的每一个;并且
任选地,响应于所述比较的结果,将核苷酸值分配给预选核苷酸位置,
其中x等于或大于2。
在一个实施方案中,方法包括将核苷酸值分配给至少2、3、5、10、20、40、50、60、70、80、90、100、200、300、400、500、600、700、800、900、或1,000个预选核苷酸位置,所述预选核苷酸位置各自独立地具有第一值,所述第一值是小于0.5、0.4、0.25、0.15、0.10、0.05、0.04、0.03、0.02、或0.01的概率的函数。
在一个实施方案中,方法包括将核苷酸值分配给至少x个核苷酸位置中的每一个,所述核苷酸位置各自独立地具有与其他x-1个第一值相比独特的第一值,并且其中所述x个第一值中的每一个都是小于0.5、0.4、0.25、0.15、0.10、0.05、0.04、0.03、0.02或0.01的概率的函数,其中x等于或大于1、2、3、5、10、20、40、50、60、70、80、90、100、200、300、400、500、600、700、800、900、或1,000。
在一个实施方案中,给至少20、40、60、80、100、120、140、160或180、200、300、400、或500个基因例如来自表1-4或图3a-4d的基因中的核苷酸位置分配核苷酸值。在一个实施方案中,将独特的第一值和/或第二值应用于分析的所述基因的至少10%、20%、30%、40%、或50%中的每一个中的受试者间隔(例如,亚基因组间隔、表达的亚基因组间隔、或两者)。
例如从以下实施方案中所看到,可应用其中优化相对大量的受试者间隔(例如,亚基因组间隔、表达的亚基因组间隔、或两者)的阈值的方法的实施方案。
在一个实施方案中,将独特的阈值应用于至少3、5、10、20、40、50、60、70、80、90、100、200、300、400、500、600、700、800、900、或1,000个不同基因中的每一个中的受试者间隔,例如,亚基因组间隔或表达的亚基因组间隔。
在一个实施方案中,给至少20、40、60、80、100、120、140、160或180、200、300、400、或500个基因例如来自表1-4或图3a-4d的基因中的核苷酸位置分配核苷酸值。在一个实施方案中,将独特的阈值应用于分析的所述基因的至少10%、20%、30%、40%、或50%中的每一个中的亚基因组间隔。
在一个实施方案中,给来自表1-4或图3a-4d的至少5、10、20、30、或40个基因中的核苷酸位置分配核苷酸值。在一个实施方案中,将独特的阈值应用于分析的所述基因的至少10%、20%、30%、40%、或50%中的每一个中的受试者间隔(例如,亚基因组间隔或表达的亚基因组间隔)。
这些和其他突变识别方法在本文其他地方更详细地讨论,例如在标题为“突变”的章节中。所述模块的要素可包括在分析肿瘤的方法中。在实施方案中,将来自标题为“突变识别”的章节的比对方法与来自标题为“比对”的章节的比对方法和/或来自标题为“诱饵”的章节的诱饵集合组合。方法可应用于来自标题为“基因选择”的章节的受试者间隔(例如,亚基因组间隔、表达的亚基因组间隔、或两者)集合。
诱饵
本文描述的方法通过适当选择诱饵(例如用于液相杂交的诱饵)以用于选择待测序的靶核苷酸,提供来自一个或多个受试者的例如来自本文描述的癌症的样品(例如,肿瘤样品)的大量基因和基因产物的优化测序。根据具有预选选择效率的诱饵集合,匹配各种受试者间隔(例如,亚基因组间隔、表达的亚基因组间隔、或两者)、或其类别的选择效率。如本章节所用,“选择效率”是指序列覆盖度的水平或深度,因为它根据靶受试者间隔(例如,亚基因组间隔、表达的亚基因组间隔、或两者)进行调整。
因此,方法(例如,以上叙述的方法的步骤(b))包括使文库与多个诱饵接触以提供选择的成员(例如,文库捕获)。
因此,在一个方面,本发明的特征在于一种分析样品的方法,所述样品例如来自癌症的肿瘤样品,例如本文描述的癌症。方法包括:
(a)获取一个或多个文库,所述一个或多个文库包括来自样品的多个成员(例如,靶成员),例如来自肿瘤样品的多个肿瘤成员;
(b)使一个或多个文库与诱饵集合(或多个诱饵集合)接触以提供选择的成员(例如文库捕获);
(c)例如通过包括测序的方法,例如使用下一代测序方法,从所述文库或文库捕获中获取来自成员(例如,肿瘤成员)的受试者间隔(例如,亚基因组间隔、表达的亚基因组间隔、或两者)的读段;
(d)通过比对方法,例如本文描述的比对方法来比对所述读段;以及
(e)将来自所述读段的核苷酸值分配(例如,使用例如贝叶斯方法或本文描述的方法的识别突变)给预选的核苷酸位置,
从而分析所述肿瘤样品,
任选地,其中方法包括使文库与多个例如至少两个、三个、四个或五个诱饵或诱饵集合接触,其中所述多个诱饵或诱饵集合中的每一个诱饵或诱饵集合具有独特(与多个诱饵集合中的其他诱饵集合完全不同)的预选选择效率。例如,每个独特的诱饵或诱饵集合都提供独特的测序深度。如本文所用,术语“诱饵集合”统称为一个诱饵或多个诱饵分子。
在一个实施方案中,方法包括获取文库,从所述文库中各自获得对应于亚基因组间隔的成员和对应于表达的基因组间隔的成员。
在一个实施方案中,方法包括获取第一文库,从所述第一文库中获得对应于亚基因组间隔的成员,并且获取第二文库,从所述第二文库中获得对应于表达的亚基因组间隔的成员。
在一个实施方案中,诱饵集合用于提供包含亚基因组间隔和表达间隔的成员或文库捕获。
在一个实施方案中,第一诱饵集合用于提供包含亚基因组间隔的成员或文库捕获,并且第二诱饵集合用于提供包含表达的亚基因组间隔的成员或文库捕获。
在一个实施方案中,多个诱饵集合中的第一诱饵集合的选择效率与多个诱饵集合中的第二诱饵集合的效率相差至少2倍。在一个实施方案中,第一诱饵集合和第二诱饵集合提供相差至少2倍的测序深度。
在一个实施方案中,方法包括使以下诱饵集合中的一个或多个与文库接触:
a)诱饵集合,其选择足够的包含亚基因组间隔的成员以提供约500x或更高的测序深度,例如对存在于不超过5%的来自样品的细胞中的突变进行测序;
b)诱饵集合,其选择足够的包含亚基因组间隔的成员以提供约200x或更高,例如约200x至约500x的测序深度,例如对存在于不超过10%的来自样品的细胞中的突变进行测序;
c)诱饵集合,其选择足够的包含亚基因组间隔的成员以提供约10-100x的测序深度,例如对一个或多个亚基因组间隔(例如,外显子)进行测序,所述一个或多个亚基因组间隔选自:i)可解释患者代谢不同药物的能力的药物基因组学(pgx)单核苷酸多态性(snp),或ii)可用于对患者进行独特地鉴定(例如,指纹分析)的基因组snp;
d)诱饵集合,其选择足够的包含受试者间隔(例如,亚基因组间隔或表达的亚基因组间隔)的成员以提供约5-50x的测序深度,例如检测结构断点,诸如基因组易位或插入缺失。例如,检测内含子断点需要5-50x的序列对跨越深度,以确保高检测可靠性。此类诱饵集合可用于检测例如易于易位/插入缺失的癌症基因;或
e)诱饵集合,其选择足够的包含受试者间隔(例如,亚基因组间隔或表达的亚基因组间隔)的成员以提供约0.1-300x的测序深度,例如检测拷贝数变化。在一个实施方案中,测序深度范围为约0.1-10x的测序深度以检测拷贝数变化。在其他实施方案中,测序深度范围为约100-300x以检测基因组snp/基因座,所述基因组snp/基因座用于评价基因组dna的拷贝数增加/减少或杂合性丢失(loh)。此类诱饵集合可用于检测例如易于扩增/缺失的癌症基因。
如本文所用的测序深度的水平(例如,x倍的测序深度水平)是指在检测和除去重复读段例如pcr重复读段后,读段(例如,独特读段)的覆盖度水平。
在一个实施方案中,诱饵集合选择含有一个或多个重排的受试者间隔(例如,亚基因组间隔或表达的亚基因组间隔),例如含有基因组重排的内含子。在此类实施方案中,诱饵集合被设计成使得重复序列被掩蔽以提高选择效率。在其中重排具有已知接合序列的那些实施方案中,互补诱饵集合可被设计成接合序列以提高选择效率。
在实施方案中,方法包括使用被设计为捕获两种或更多种不同的靶标分类的诱饵,每种分类都具有不同的诱饵设计策略。在实施方案中,本文公开的杂交捕获方法和组合物捕获靶序列(例如,靶成员)的确定子集并且提供靶序列的均匀覆盖,同时最小化所述子集之外的覆盖。在一个实施方案中,靶序列包含基因组dna中的整个外显子组、或其选择的子集。在另一个实施方案中,靶序列包含较大的染色体区域,例如整个染色体臂。本文公开的方法和组合物提供不同的诱饵集合,以用于实现复杂靶核酸序列(例如,核酸文库)的不同覆盖深度和模式。
在一个实施方案中,方法包括提供一个或多个核酸文库的选择的成员(例如,文库捕获)。方法包括:
提供一个或多个文库(例如,一个或多个核酸文库),所述一个或多个文库包括多个成员,例如靶核酸成员(例如,包括多个肿瘤成员、参考成员和/或pgx成员);
例如在基于液相的响应中,使一个或多个文库与多个诱饵(例如,寡核苷酸诱饵)接触以形成包含多个诱饵/成员杂交体的杂交混合物;
例如通过使所述杂交混合物与允许分离所述多个诱饵/成员杂交体的结合实体接触,将多个诱饵/成员杂交体与所述杂交混合物分离,
从而提供文库捕获(例如,来自一个或多个文库的选择或富集的核酸分子子集),
任选地,其中多个诱饵包括以下的两种或更多种:
a)第一诱饵集合,其选择高水平靶标(例如,一个或多个肿瘤成员,所述一个或多个肿瘤成员包含受试者间隔(例如,亚基因组间隔或表达的亚基因组间隔),诸如基因、外显子或碱基),对于所述高水平靶标,需要最深覆盖以实现对于以低频率例如约5%或更少(即,来自样品的细胞中的5%在其基因组中具有改变)出现的改变(例如,一个或多个突变)的高水平灵敏度。在一个实施方案中;第一诱饵集合选择(例如,互补于)包含改变(例如,点突变)的肿瘤成员,所述改变需要约500x或更高的测序深度;
b)第二诱饵集合,其选择中等水平靶标(例如,一个或多个肿瘤成员,所述一个或多个肿瘤成员包含受试者间隔(例如,亚基因组间隔或表达的亚基因组间隔),诸如基因、外显子或碱基),对于所述中等水平靶标,需要高覆盖度以实现对于以比a)中的高水平靶标更高的频率例如约10%的频率(即,来自样品的细胞中的10%在其基因组中具有改变)出现的改变(例如,一个或多个突变)的高水平灵敏度。在一个实施方案中;第二诱饵集合选择(例如,互补于)包含改变(例如,点突变)的肿瘤成员,所述改变需要约200x或更高的测序深度;
c)第三诱饵集合,其选择低水平靶标(例如,一个或多个pgx成员,所述一个或多个pgx成员包含受试者间隔(例如,亚基因组间隔或表达的亚基因组间隔),诸如基因、外显子或碱基),对于所述低水平靶标需要低至中等覆盖度以实现例如对检测杂合等位基因的高水平灵敏度。例如,杂合等位基因的检测需要10-100x的测序深度以确保高检测可靠性。在一个实施方案中,第三诱饵集合选择一个或多个受试者间隔(例如,亚基因组间隔、表达的亚基因组间隔、或两者,例如外显子),所述一个或多个受试者间隔选自:a)可解释患者代谢不同药物的能力的药物基因组学(pgx)单核苷酸多态性(snp),或b)可用于对患者进行独特地鉴定(例如,指纹分析)的基因组snp;
d)第四诱饵集合,其选择第一内含子靶标(例如,包含内含子序列的成员),对于所述第一内含子靶标需要低至中等覆盖度以例如检测结构断点,诸如基因组易位或插入缺失。例如,检测内含子断点需要5-50x的序列对跨越深度,以确保高检测可靠性。所述第四诱饵集合可用于检测例如易于易位/插入缺失的癌症基因;或
e)第五诱饵集合,其选择第二内含子靶标(例如,内含子成员),对于所述第二内含子靶标需要稀疏的覆盖以提高检测拷贝数变化的能力。例如,检测若干个末端外显子的一拷贝缺失需要0.1-300x覆盖度以确保高检测可靠性。在一个实施方案中,覆盖深度范围为约0.1-10x以检测拷贝数变化。在其他实施方案中,覆盖深度范围为约100-300x以检测基因组snp/基因座以评价基因组dna的拷贝数增加/减少或杂合性丢失(loh)。所述第五诱饵集合可用于检测例如易于扩增/缺失的癌症基因。
可使用上述诱饵集合中的两个、三个、四个或更多个的任何组合,例如,第一诱饵集合和第二诱饵集合的组合;第一诱饵集合和第三诱饵集合;第一诱饵集合和第四诱饵集合;第一诱饵集合和第五诱饵集合;第二诱饵集合和第三诱饵集合;第二诱饵集合和第四诱饵集合;第二诱饵集合和第五诱饵集合;第三诱饵集合和第四诱饵集合;第三诱饵集合和第五诱饵集合;第四诱饵集合和第五诱饵集合;第一诱饵集合、第二诱饵集合和第三诱饵集合;第一诱饵集合、第二诱饵集合和第四诱饵集合;第一诱饵集合、第二诱饵集合和第五诱饵集合;第一诱饵集合、第二诱饵集合、第三诱饵集合和第四诱饵集合;第一诱饵集合、第二诱饵集合、第三诱饵集合、第四诱饵集合和第五诱饵集合等。
在一个实施方案中,第一诱饵集合、第二诱饵集合、第三诱饵集合、第四诱饵集合或第五诱饵集合中的每一个都具有预选的选择(例如,捕获)效率。在一个实施方案中,针对根据a)-e)的所有五个诱饵中的至少两个、三个、四个的选择效率的值是相同的。在其他实施方案中,针对根据a)-e)的所有五个诱饵中的至少两个、三个、四个的选择效率的值是不同的。
在一些实施方案中,至少两个、三个、四个或所有五个诱饵集合具有不同的预选效率值。例如,选择效率的值选自以下的一种或多种:
(i)第一预选效率,具有至少约500x或更高的测序深度的第一选择效率的值(例如,具有大于第二、第三、第四或第五预选选择效率的选择效率的值(例如,比第二选择效率的值大约2-3倍;比第三选择效率的值大约5-6倍;比第四选择效率的值大约10倍;比第五选择效率的值大约50至5,000倍);
(ii)第二预选效率,具有至少约200x或更高的测序深度的第二选择效率的值,例如具有大于第三、第四或第五预选选择效率的选择效率的值(例如,比第三选择效率的值大约2倍;比第四选择效率的值大约4倍;比第五选择效率的值大约20至2,000倍);
(iii)第三预选效率,具有至少约100x或更高的测序深度的第三选择效率的值,例如具有大于第四或第五预选选择效率的选择效率的值(例如,比第四选择效率的值大约2倍;比第五选择效率的值大约10至1000倍);
(iv)第四预选效率,具有至少约50x或更高的测序深度的第四选择效率的值,例如具有大于第五预选选择效率的选择效率的值(例如,比第五选择效率的值大约50至500倍);或
(v)第五预选效率,具有至少约10x至0.1x的测序深度的第五选择效率的值。
在某些实施方案中,选择效率的值通过以下的一种或多种来修改:不同诱饵集合的差异表示、诱饵子集的差异重叠、差异诱饵参数、不同诱饵集合的混合、和/或使用不同类型的诱饵集合。例如,选择效率(例如,每个诱饵集合/靶分类的相对序列覆盖度)的变化可通过改变以下的一种或多种来调整:
(i)不同诱饵集合的差异表示–捕获给定靶标(例如,靶成员)的诱饵集合设计可包括在更多/更少数量的拷贝中,以增强/减少相对靶标覆盖深度;
(ii)诱饵子集的差异重叠–捕获给定靶标(例如,靶成员)的诱饵集合设计可包括相邻诱饵之间更长或更短的重叠,以增强/减少相对靶标覆盖深度;
(iii)差异诱饵参数–捕获给定靶标(例如,靶成员)的诱饵集合设计可包括序列修饰/长度更短以减小捕获效率并且降低相对靶标覆盖深度;
(iv)不同诱饵集合的混合–设计为捕获不同靶标集合的诱饵集合可在不同摩尔比下混合,以增强/减少相对靶标覆盖深度;
(v)使用不同类型的寡核苷酸诱饵集合–在某些实施方案中,诱饵集合可包括:
(a)一个或多个化学(例如,非酶促)合成的(例如,单独合成的)诱饵,
(b)一个或多个以阵列形式合成的诱饵,
(c)一个或多个酶促制备的,例如体外转录的诱饵;
(d)(a)、(b)和/或(c)的任何组合,
(e)一个或多个dna寡核苷酸(例如,天然或非天然存在的dna寡核苷酸),
(f)一个或多个rna寡核苷酸(例如,天然或非天然存在的rna寡核苷酸),
(g)(e)和(f)的组合,或
(h)上述任何一项的组合。
不同的寡核苷酸组合可在不同比率下混合,例如选自1:1、1:2、1:3、1:4、1:5、1:10、1:20、1:50、1:100、1:1000等的比率。在一个实施方案中,化学合成的诱饵与阵列产生的诱饵的比率选自1:5、1:10或1:20。dna或rna寡核苷酸可以是天然或非天然存在的。在某些实施方案中,诱饵包含一个或多个非天然存在的核苷酸,以例如增加熔解温度。示例性非天然存在的寡核苷酸包括修饰的dna或rna核苷酸。示例性修饰的核苷酸(例如,修饰的rna或dna核苷酸)包括但不限于锁核酸(lna),其中lna核苷酸的核糖部分用连接2'氧和4'碳的额外桥修饰;肽核酸(pna),例如由通过肽键连接的重复n-(2-氨乙基)-甘氨酸单元组成的pna;修饰以捕获低gc区域的dna或rna寡核苷酸;双环核酸(bna);交联寡核苷酸;修饰的5-甲基脱氧胞苷;以及2,6-二氨基嘌呤。其他修饰的dna和rna核苷酸是本领域中已知的。
在某些实施方案中,获得靶序列(例如,靶成员)的基本上均一或均匀的覆盖。例如,在每个诱饵集合/靶分类中,可通过修改诱饵参数来优化覆盖的均一性,例如,通过以下的一种或多种:
(i)增加/减少诱饵表示或重叠可用于增强/减少靶标(例如,靶成员)的覆盖度,所述靶标相对于相同分类中的其他靶标是欠覆盖/过度覆盖;
(ii)对于低覆盖度,难以捕获靶序列(例如,高gc含量序列),扩大用诱饵集合靶向的区域以覆盖例如相邻序列(例如,富含gc较少的相邻序列);
(iii)修饰诱饵序列可用于减少诱饵的二级结构并且提高其选择效率;
(iv)修改诱饵长度可用于均衡相同分类内不同诱饵的熔解杂交动力学。诱饵长度可直接修改(通过产生具有不同长度的诱饵)或间接修改(通过产生长度一致的诱饵,并且用任意序列替换诱饵端部);
(v)修饰针对相同靶区域具有不同取向的诱饵(即正向和反向链)可具有不同的结合效率。可选择具有为每个靶标提供最佳覆盖度的任一取向的诱饵集合;
(vi)修改存在于每个诱饵上的结合实体例如捕获标签(例如生物素)的量可影响其结合效率。增加/减少靶向特异性靶标的诱饵的标签水平可用于增强/减少相对靶标覆盖度;
(vii)修改用于不同诱饵的核苷酸类型可用于影响对靶标的结合亲和力,并且增强/减少相对靶标覆盖度;或
(viii)使用例如具有更稳定的碱基配对的修饰的寡核苷酸诱饵,可用于均衡低gc含量或正常gc含量的区域相对于高gc含量的区域之间的熔解杂交动力学。
例如,可使用不同类型的寡核苷酸诱饵集合。
在一个实施方案中,通过使用不同类型的诱饵寡核苷酸以涵盖预选的靶区域来修改选择效率的值。例如,第一诱饵集合(例如,包括10,000-50,000个rna或dna诱饵的基于阵列的诱饵集合)可用于覆盖较大的靶区域(例如,总共1-2mb的靶区域)。第一诱饵集合可掺有第二诱饵集合(例如,包括小于5,000个诱饵的单独合成的rna或dna诱饵集合)以覆盖预选的靶区域(例如,选择的靶区域的跨越例如250kb或更少的所关注的亚基因组间隔)和/或具有更高二级结构例如更高gc含量的区域。选择的所关注的受试者间隔(例如,亚基因组间隔、表达的亚基因组间隔、或两者)可对应于本文描述的基因或基因产物、或其片段中的一种或多种。根据所需的诱饵重叠,第二诱饵集合可包括约1-5,000、2-5,000、3-5,000、10-5,000、100-5,000、500-5,000、100-5,000、1,000-5,000、2,000-5,000个诱饵。在其他实施方案中,第二诱饵集合可包括掺入到第一诱饵集合中的选择的寡聚诱饵(例如,小于400、200、100、50、40、30、20、10、5、4、3、2或1个诱饵)。第二诱饵集合可在单个寡聚诱饵的任何比率下混合。例如,第二诱饵集合可包括呈1:1等摩尔比存在的单个诱饵。可替代地,第二诱饵集合可包括在不同比率(例如,1:5、1:10、1:20)下存在的单个诱饵,例如以优化某些靶标(例如,与其他靶标相比,某些靶标可具有5-10x的第二诱饵集合)的捕获。
在其他实施方案中,参考当使用等摩尔诱饵混合物,然后相对于第二组诱饵将过量的差异第一组诱饵引入整个诱饵混合物时观察到的差异序列捕获效率,通过调整诱饵的相对丰度或结合实体的密度(例如,半抗原或亲和标签密度),通过平衡一个组(例如,第一、第二或第三多个诱饵)内的单个诱饵的效率来调整选择效率。
在一个实施方案中,方法包括使用多个诱饵集合,所述多个诱饵集合包括从肿瘤细胞(在本文中又称为“肿瘤诱饵集合”)中选择肿瘤成员例如包含受试者间隔(例如,亚基因组间隔或表达的亚基因组间隔)的核酸分子的诱饵集合。肿瘤成员可以是存在于肿瘤细胞中的任何核苷酸序列,例如存在于肿瘤或癌症细胞中的如本文所述的突变核苷酸序列、野生型核苷酸序列、pgx核苷酸序列、参考核苷酸序列或内含子核苷酸序列。在一个实施方案中,肿瘤成员包含以低频率(例如,来自肿瘤样品的细胞中的约5%或更少在其基因组中具有改变)出现的改变(例如,一个或多个突变)。在其他实施方案中,肿瘤成员包含以来自肿瘤样品的细胞中的约10%的频率出现的改变(例如,一个或多个突变)。在其他实施方案中,肿瘤成员包含来自存在于肿瘤细胞中的pgx基因或基因产物、内含子序列(例如,本文所述的内含子序列)、参考序列的亚基因组间隔。
另一方面,本发明的特征在于本文描述的诱饵集合、本文描述的单个诱饵集合的组合,例如本文描述的组合。诱饵集合可以是试剂盒的一部分,所述试剂盒可任选地包括说明书、标准品、缓冲剂或酶或其他试剂。
基因选择
本文描述了预选的受试者间隔(例如,亚基因组间隔、表达的亚基因组间隔、或两者),以用于分析例如基因和其他区域的集合或组的亚基因组间隔的组或集合。
因此,在实施方案中,方法包括例如通过下一代测序方法,对受试者间隔(例如,亚基因组间隔或表达的亚基因组间隔)进行测序,所述受试者间隔来自获取的核酸样品的至少5、6、7、8、9、10、15、20、25、30、40、50、60、70、80、90、100、200、300、400、500或更多个基因或基因产物,其中基因或基因产物选自表1-4或图3a-4d,从而分析例如来自本文描述的癌症的肿瘤样品。
因此,在一个方面,本发明的特征在于一种分析样品的方法,所述样品例如来自恶性(或恶化前)血液病的肿瘤样品,例如本文描述的恶性(或恶化前)血液病。方法包括:
(a)获取一个或多个文库,所述一个或多个文库包括来自样品的多个成员,例如来自肿瘤样品的多个肿瘤成员,所述肿瘤样品来自恶性(或恶化前)血液病,例如本文描述的恶性(或恶化前)血液病;
(b)任选地,例如通过使一个或多个文库与诱饵集合(或多个诱饵集合)接触以提供选择的成员(例如文库捕获),使一个或多个文库富含预选序列;
(c)例如通过包括测序的方法,例如使用下一代测序方法,从所述文库或文库捕获中获取来自成员例如肿瘤成员的受试者间隔(例如,亚基因组间隔或表达的亚基因组间隔)的读段;
(d)通过比对方法,例如本文描述的比对方法来比对所述读段;以及
(e)将来自所述读段的核苷酸值分配(例如,使用例如贝叶斯方法或本文描述的方法的识别突变)给预选的核苷酸位置,从而分析所述肿瘤样品,
任选地,其中方法包括例如通过下一代测序方法,对受试者间隔(例如,亚基因组间隔或表达的亚基因组间隔)进行测序,所述受试者间隔来自样品的至少5、6、7、8、9、10、15、20、25、30、40、50、60、70、80、90、100、200、300、400、500或更多个基因或基因产物,其中基因或基因产物选自表1-4或图3a-4d。
在一个实施方案中,存在步骤(b)。在一个实施方案中,不存在步骤(b)。
在另一个实施方案中,分析以下集合或组中的一种的受试者间隔(例如,亚基因组间隔、表达的亚基因组间隔、或两者)。例如,与肿瘤或癌症基因或基因产物、参考(例如,野生型)基因或基因产物以及pgx基因或基因产物相关的受试者间隔(例如,亚基因组间隔、表达的亚基因组间隔、或两者)可提供来自肿瘤样品的亚基因组间隔的组或集合。
在一个实施方案中,方法从肿瘤样品中获取例如序列、受试者间隔(例如,亚基因组间隔、表达的亚基因组间隔、或两者)集合的读段,其中受试者间隔(例如,亚基因组间隔、表达的亚基因组间隔、或两者)选自以下的至少1、2、3、4、5、6、7种或所有:
a)来自根据表1-4或图3a-4d的突变或野生型基因或基因产物的至少5、6、7、8、9、10、15、20、25、30、40、50、60、70、80、90、100、200、300、400、500或更多个受试者间隔,例如亚基因组间隔、或表达的亚基因组间隔、或两者;
b)来自与肿瘤或癌症相关的基因或基因产物(例如,作为肿瘤或癌症的阳性或阴性治疗响应预测因子、作为阳性或阴性预后因素、或能实现肿瘤或癌症的鉴别诊断,例如,根据表1-4或图3a-4d的基因或基因产物)的至少5、6、7、8、9、10、15、20、25、30、40、50、60、70、80、90、100、200、300、400、500或更多个受试者间隔(例如,亚基因组间隔、表达的亚基因组间隔、或两者);
c)来自具有一种亚基因组间隔的突变或野生型基因或基因产物(例如,单核苷酸多态性(snp))的至少5、6、7、8、9、10、15、20、25、30、40、50、60、70、80、90、100、200、300、400、500或更多个受试者间隔(例如,亚基因组间隔、表达的亚基因组间隔、或两者),所述亚基因组间隔存在于与药物代谢、药物响应性或毒性中的一种或多种相关的基因或基因产物(在本文中又称为“pgx”基因)中,所述基因或基因产物选自表1-4或图3a-4d;
d)来自具有一种受试者间隔(例如,亚基因组间隔或表达的亚基因组间隔)的突变或野生型pgx基因或基因产物(例如,单核苷酸多态性(snp))的至少5、6、7、8、9、10、15、20、25、30、40、50、60、70、80、90、100、200、300、400、500或更多个受试者间隔(例如,亚基因组间隔、表达的亚基因组间隔、或两者),所述受试者间隔存在于选自表1-4或图3a-4d的基因或基因产物中,所述基因或基因产物与以下的一种或多种相关:(i)用药物治疗的癌症患者的存活率更高(例如,用紫杉醇治疗的乳腺癌患者的存活率更高);(ii)紫杉醇代谢;(iii)对药物的毒性;或(iv)对药物的副作用;
e)涉及根据表1-4或图3a-4d的至少5、6、7、8、9、10、15、20、25、30、40、50、60、70、80、90、100、200、300、400、500或更多个基因或基因产物的多个易位改变;
f)至少五个选自表1-4或图3a-4d的基因或基因产物,其中例如在预选位置处的等位变异与预选类型的肿瘤相关,并且其中所述等位变异存在于所述肿瘤类型的小于5%的细胞中;
g)至少五个选自表1-4或图3a-4d的基因或基因产物,所述基因或基因产物包埋在富含gc的区域中;或
h)至少五个表示得癌症的遗传(例如,种系风险)因素的基因或基因产物(例如,基因或基因产物选自表1-4或图3a-4d)。
在另一个实施方案中,方法从肿瘤样品中获取例如序列、受试者间隔(例如,亚基因组间隔、表达的亚基因组间隔、或两者)集合的读段,其中受试者间隔(例如,亚基因组间隔、表达的亚基因组间隔、或两者)选自表1中描述的5、6、7、8、9、10、15、20、25、30、40、50、60、70、80、90、100、200、300、400个或所有的基因或基因产物。
在另一个实施方案中,方法从肿瘤样品中获取例如序列,受试者间隔(例如,亚基因组间隔、表达的亚基因组间隔、或两者)集合的读段,其中受试者间隔(例如,亚基因组间隔、表达的亚基因组间隔、或两者)选自表2中描述的5、6、7、8、9、10、15、20、25、30个或所有的基因或基因产物。
在另一个实施方案中,方法从肿瘤样品中获取例如序列,受试者间隔(例如,亚基因组间隔、表达的亚基因组间隔、或两者)集合的读段,其中受试者间隔(例如,亚基因组间隔、表达的亚基因组间隔、或两者)选自表3中描述的5、6、7、8、9、10、15、20、25、30、40、50、60、70、80、90、100、200、300个或所有的基因或基因产物。
在另一个实施方案中,方法从肿瘤样品中获取例如序列,受试者间隔(例如,亚基因组间隔、表达的亚基因组间隔、或两者)集合的读段,其中受试者间隔(例如,亚基因组间隔、表达的亚基因组间隔、或两者)选自表4中描述的5、6、7、8、9、10、15、20、25、30、40、50、60、70、80个或所有的基因或基因产物。
这些和其他亚基因组间隔的集合和组在本文其他地方更详细地讨论,例如在标题为“基因选择”的章节中。
本文描述的任何方法可与以下实施方案中的一种或多种组合。
在其他实施方案中,样品是肿瘤样品,例如包含一个或多个恶化前或恶性细胞。在某些实施方案中,样品例如肿瘤样品从恶性(或恶化前)血液病,例如本文描述的恶性(或恶化前)血液病中获取。在某些实施方案中,样品例如肿瘤样品从实体瘤、软组织肿瘤或转移病变中获取。在其他实施方案中,样品例如肿瘤样品包含来自手术切缘的组织或细胞。在某些实施方案中,样品例如肿瘤样品包含肿瘤浸润淋巴细胞。样品可以是组织学正常的组织。在另一个实施方案中,样品例如肿瘤样品包含一个或多个循环肿瘤细胞(ctc)(例如,从血液样品中获取的ctc)。在一个实施方案中,样品例如肿瘤样品包含一个或多个非恶性细胞。在一个实施方案中,样品例如肿瘤样品包含一个或多个肿瘤浸润淋巴细胞。
在一个实施方案中,方法还包括获取样品,例如本文所述的肿瘤样品。样品可以直接或间接地获取。在一个实施方案中,例如通过分离或纯化,从包含恶性细胞和非恶性细胞(例如,肿瘤浸润淋巴细胞)的样品中获取样品。
在其他实施方案中,方法包括使用本文描述的方法评估样品,例如组织学正常的样品,所述样品例如来自手术切缘。申请人发现从组织学正常的组织(例如,或者组织学正常的组织边缘)中获得的样品仍然可具有如本文所述的改变。因此,方法还可包括基于检测到的改变的存在来重新分类组织样品。
在另一个实施方案中,被获取或分析的读段中的至少10%、20%、30%、40%、50%、60%、70%、80%或90%是针对来自本文描述的基因,例如来自表1-4或图3a-4d的基因的受试者间隔(例如,亚基因组间隔、表达的亚基因组间隔、或两者)。
在一个实施方案中,在方法中进行的突变识别中的至少10%、20%、30%、40%、50%、60%、70%、80%或90%是针对来自本文描述的基因或基因产物,例如来自表1-4或图3a-4d的基因或基因产物的受试者间隔(例如,亚基因组间隔、表达的亚基因组间隔、或两者)。
在一个实施方案中,方法使用的独特阈值中的至少10%、20%、30%、40%、50%、60%、70%、80%或90%是针对来自本文描述的基因或基因产物,例如来自表1-4或图3a-4d的基因或基因产物的受试者间隔(例如,亚基因组间隔、表达的亚基因组间隔、或两者)。
在一个实施方案中,注释的或向第三方报告的突变识别中的至少10%、20%、30%、40%、50%、60%、70%、80%或90%是针对来自本文描述的基因或基因产物,例如来自表1-4或图3a-4d的基因或基因产物的受试者间隔(例如,亚基因组间隔、表达的亚基因组间隔、或两者)。
在一个实施方案中,方法包括获取从肿瘤和/或对照核酸样品(例如,ffpe衍生的核酸样品)中获得的核苷酸序列读段。
在一个实施方案中,通过ngs测序方法来提供读段。
在一个实施方案中,方法包括提供核酸成员的一个或多个文库,并且对来自所述一个或多个文库的多个成员的预选亚基因组间隔进行测序。在实施方案中,方法可包括选择所述一个或多个文库的子集以用于测序的步骤,例如基于液相的选择或基于固相载体(例如,阵列)的选择。
在一个实施方案中,方法包括使一个或多个文库与多个诱饵接触以提供选择的核酸亚组(例如,文库捕获)的步骤。在一个实施方案中,接触步骤在液相杂交中实现。在另一个实施方案中,接触步骤在固相载体例如阵列中实现。在某些实施方案中,方法包括通过另外的一轮或多轮杂交来重复杂交步骤。在一些实施方案中,方法还包括使文库捕获受到另外的一轮或多轮与相同或不同诱饵集合(collection)进行的杂交。
在其他实施方案中,方法还包括分析文库捕获。在一个实施方案中,通过测序方法,例如本文所述的下一代测序方法来分析文库捕获。方法包括通过例如液相杂交来分离文库捕获,并且通过核酸测序对文库捕获进行处理(subject)。在某些实施方案中,可重新测序文库捕获。下一代测序方法是本领域中已知的,并且描述于例如metzker,m.(2010)naturebiotechnologyreviews11:31-46中。
在一个实施方案中,核苷酸位置的分配值被传输至第三方,任选地,具有解释性注释。
在一个实施方案中,核苷酸位置的分配值不被传输至第三方。
在一个实施方案中,多个核苷酸位置的分配值被传输至第三方,任选地,具有解释性注释,并且第二多个核苷酸位置的分配值不被传输至第三方。
在一个实施方案中,对至少0.01、0.02、0.03、0.04、0.05、0.1、0.2、0.3、0.4、0.5、0.6、0.7、0.8、0.9、1.0、2.0、5.0、10、15或30个兆碱基,例如基因组碱基进行测序。
在一个实施方案中,方法包括评估多个读段,所述多个读段包含至少一个snp。
在一个实施方案中,方法包括确定样品和/或对照读段中的snp等位基因比率。
在一个实施方案中,方法包括例如通过条形码解卷积,将一个或多个读段分配给受试者。
在一个实施方案中,方法包括例如通过条形码解卷积,将一个或多个读段分配为肿瘤读段或对照对数。
在一个实施方案中,方法包括例如通过与参考序列比对,作图所述一个或多个读段中的每一个。
在一个实施方案中,方法包括记忆被识别的突变。
在一个实施方案中,方法包括注释被识别的突变,例如用突变结构的指示来注释被识别的突变,例如错义突变或功能,例如疾病表型。
在一个实施方案中,方法包括获取肿瘤和对照核酸的核苷酸序列读段。
在一个实施方案中,方法包括例如使用贝叶斯识别方法或非贝叶斯识别方法,针对受试者间隔(例如,亚基因组间隔、表达的亚基因组间隔、或两者)中的每一个识别核苷酸值,例如变体,例如突变。
在一个实施方案中,同时处理例如来自不同受试者的多个样品。
本文公开的方法可用于检测存在于受试者的基因组或转录组中的改变,并且可应用于dna和rna测序,例如靶向的rna和/或dna测序。因此,本发明特征的另一方面包括用于靶向rna测序以检测本文描述的改变的方法,例如对源自rna的cdna的测序,所述rna从样品,例如ffpe样品、血液样品或骨髓抽吸物样品中获取。改变可以是重排,例如编码基因融合体的重排。在其他实施方案中,方法包括检测基因或基因产物水平的变化(例如,增加或减少),例如本文描述的基因或基因产物的表达变化。方法可任选地包括使样品富含靶rna的步骤。在其他实施方案中,方法包括使样品耗尽某些高丰度rna,例如核糖体或珠蛋白rna的步骤。rna测序方法可单独使用或与本文描述的dna测序方法组合使用。在一个实施方案中,方法包括执行dna测序步骤和rna测序步骤。可按任何顺序执行方法。例如,方法可包括通过rna测序来确认本文描述的改变的表达,例如确认通过本发明的dna测序方法检测到的突变或融合体的表达。在其他实施方案中,方法包括执行rna测序步骤,然后执行dna测序步骤。
另一方面,本发明的特征在于一种方法,所述方法包括针对靶向的亚基因组区域构建测序/比对假象(artifact)的数据库。在一个实施方案中,数据库可用于过滤掉虚假突变识别并且提高特异性。在一个实施方案中,通过对不相关的非肿瘤(例如,ffpe、血液或骨髓抽吸物)样品或细胞系进行测序,并且记录比预期更频繁出现的非参考等位基因事件来构建数据库,所述非参考等位基因事件由于在这些正常样品中的一个或多个中单独的随机测序错误而引起。这种方法可将种系变异分类为假象,但这在涉及体细胞突变的方法中是可接受的。如果需要,可通过针对已知的种系变异(除去常见变体)和针对仅出现在1个个体中的假象(除去罕见变异)对这个数据库进行过滤,改善将种系变异作为假象的这种错误分类。
当例如应用于癌症相关的基因组区段时,本文公开的方法允许合并许多优化的要素,所述优化的要素包括优化的基于诱饵的选择、优化的比对和优化的突变识别。本文描述的方法提供基于ngs的肿瘤分析,其可在癌症、基因和位点的基础上进行优化。这可例如应用于本文描述的基因/位点和肿瘤类型。方法使用给定的测序技术来优化突变检测的灵敏度和特异性水平。癌症、基因和位点的优化提供了非常高水平的灵敏度/特异性(例如,针对这两者,>99%),所述非常高水平的灵敏度/特异性是临床产品必不可少的。
本文描述的方法从常规真实的样品中使用下一代测序技术,针对合理可操作基因(所述基因的范围可通常为50至500个基因)的全面集合提供临床和监管级的全面分析和基因组畸变的解释,以便告知最佳治疗和疾病管理决策。
本文描述的方法为肿瘤学家/病理学家提供一站式地发送肿瘤样品并且接收针对所述肿瘤的基因组和其他分子变化的全面分析和描述,以便告知最佳治疗和疾病管理决策。
本文描述的方法提供了一种稳健、真实的临床肿瘤学诊断工具,其采用标准的可用肿瘤样品,并且在一个测试中提供全面基因组和其他分子畸变分析以向肿瘤学家提供对何种畸变可驱动肿瘤的全面描述,并且可用于告知肿瘤学家治疗决策。
本文描述的方法提供具有临床级质量的对患者癌症基因组的全面分析。方法包括最相关的基因和潜在的改变,并且包括以下的一种或多种:对突变(例如,插入缺失或碱基取代)、拷贝数、重排(例如,易位)、表达和表观遗传学标记物的分析。可使用对可操作结果的描述性报告,将遗传分析的输出置于上下文中研究。方法结合使用一套最新的相关科学和医学知识。
本文描述的方法提供增加患者护理的质量和效率。这包括以下应用:其中肿瘤属于罕见或很少研究的类型,使得没有护理标准或患者对已建立的治疗线难以治愈,并且选择进一步治疗或临床试验参与的合理依据可以是有用的。例如,方法允许在任何治疗点时选择,其中肿瘤学家将通过具有可用于告知决策的完整“分子图像”和/或“分子亚诊断”来受益。
本文描述的方法可包括例如以电子、基于网络或纸质的形式,向患者或向另一个人或主体、例如护理人员、例如医生、例如肿瘤学家、医院、诊所、第三方付款人、保险公司或政府办公室提供报告。例如针对与样品类型的肿瘤相关的受试者间隔(例如,亚基因组间隔、表达的亚基因组间隔、或两者),报告可包括来自方法的输出,例如核苷酸值的鉴定;改变、突变或野生型序列存在或不存在的指示。报告还可包括关于序列,例如改变、突变或野生型序列在疾病中的作用的信息。此类信息可包括关于预后、耐药性或潜在或建议的治疗选项的信息。报告可包括关于治疗选项的可能有效性、治疗选项的可接受性或将治疗选项应用于患者的可取性的信息,所述患者例如具有在测试中鉴定并且在实施方案中,在报告中鉴定的序列、改变的患者。例如,报告可包括关于例如与其他药物组合向患者进行的药物施用,例如在预选剂量下或在预选治疗方案中的施用的信息或建议。在一个实施方案中,并非在报告中鉴定了在方法中鉴定的所有突变。例如,报告可限于具有预选水平的与例如使用预选治疗选项治疗的癌症的发生、预后、时期或易感性相关的基因中的突变。本文特征的方法允许距离由实施方法的主体接收样品后的7天、14天或21天内,例如向本文描述的主体传递报告。
因此,本发明特征的方法允许例如在接收样品后的7天、14天或21天内的快速周转时间。
本文描述的方法也可用于评估组织学正常的样品,例如来自手术切缘的样品。如果检测到如本文所述的一个或多个改变,则可将组织重新分类为例如恶性或恶化前的,和/或可修改疗程。
在某些方面,本文描述的测序方法可用于非癌症应用,例如法医学应用(例如,作为使用牙科记录的替代或补充的鉴定)、亲子鉴定、以及疾病诊断和预后,例如以用于传染性疾病、自身免疫性病症、囊性纤维化、亨廷顿氏病、阿尔茨海默病等。例如,通过本文描述的方法鉴定遗传改变可表示个体存在特定病症或个体发展特定病症的风险。
除非另有定义,否则本文使用的所有技术和科学术语具有与本发明所属领域中的普通技术人员所通常理解的相同的意义。尽管可在本发明的实践或测试中使用与本文描述的那些方法和材料类似或等效的方法和材料,但以下描述了合适的方法和材料。本文提到的所有出版物、专利申请、专利和其他参考文献以引用的方式整体并入。另外,材料、方法和实施例仅是说明性的并非意图为限制性的。
本发明的其他特征和优点将根据以下具体实施方式、附图以及根据权利要求书而显而易知。
附图说明
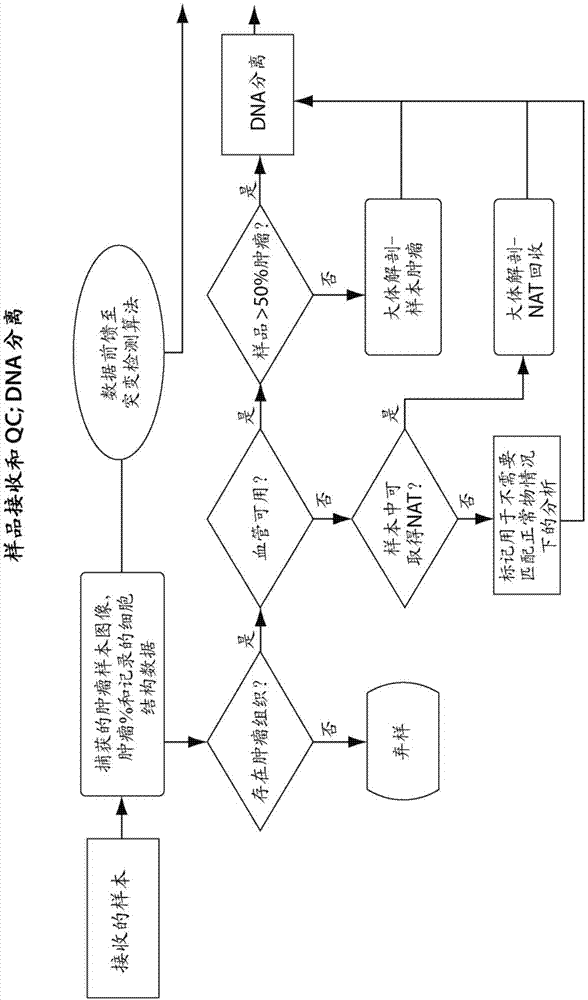
图1a-1f示出了用于肿瘤样品的多基因分析的方法的实施方案的流程图描绘。
图2描绘了先验期望值和读取深度对突变检测的影响。
图3a-3b描绘了根据本文描述的方法可评估的另外示例性基因(例如,在实体瘤中)。
图4a-4d描绘了根据本文描述的方法可评估的另外示例性基因(例如,在恶性血液病或肉瘤中)。
图5-6描绘了示出全外显子组突变负荷与从靶向基因测量的突变负荷之间的相关性的散点图。
图7a-7d描绘了肺癌中的肿瘤突变负荷分布。通过分别在10,676例肺腺癌(图7a)、1,960例肺鳞状细胞癌(图7b)、220例肺大细胞癌(图7c)以及784例肺小细胞癌临床样本(图7d)中进行全面基因组谱分析来确定tmb。
图8a-8e描绘了肺癌中的遗传改变流行率(prevalence)。通过全面基因组谱分析,分别鉴定了频繁交替出现在肺腺癌(图8a)、肺鳞状细胞癌(图8b)、肺大细胞癌(图8c)以及肺小细胞癌(图8d)中的二十五个基因。示出了肺癌的所有四种亚型的聚集基因流行率(图8e)。sv:短变体;cna:拷贝数改变;re:重排;多个:相同基因中的多种类型的改变。
图9a-9b描述了结直肠腺癌中的肿瘤突变负荷分布。通过分别在6,742例结肠腺癌(图9a)和1,176例直肠腺癌临床样本(图9b)中进行全面基因组谱分析来确定tmb。
图10a-10c描绘了结直肠腺癌中的遗传改变流行率。通过全面基因组谱分析,分别鉴定了频繁交替出现在结肠腺癌(图10a)和直肠腺癌(图10b)中的二十五个基因。示出了结直肠腺癌的聚集基因流行率(图10c)。sv:短变体;cna:拷贝数改变;re:重排;多个:相同基因中的多种类型的改变。
图11描绘了二十四种类型的肿瘤中的肿瘤突变负荷分布。通过在总共15508例临床样本中进行全面基因组谱分析来确定tmb,所述临床样本包括例如膀胱、脑、乳腺、子宫颈、头颈部、肝脏、卵巢、胰腺、前列腺、皮肤、胃和子宫的肿瘤。
具体实施方式
本发明至少部分地基于以下发现:例如使用基于杂交捕获的下一代测序(ngs)平台对来自患者样品的一小部分基因组或外显子组谱分析,起到有效替代总突变负荷分析的作用。
不受理论约束,据信产生免疫原性肿瘤新抗原的可能性随着突变发展以概率方式增加,增加了免疫识别的可能性(gubin和schreiber.science350:158-9,2015)。然而,评估总突变负荷需要全外显子组测序(wes)。这种方法需要专门的组织处理、匹配的正常样本,并且目前主要作为研究工具执行。鉴于在临床环境中执行wes的技术和信息学挑战,需要检测突变负荷的替代方法。本文描述的包括验证的基于杂交捕获的ngs平台的方法具有若干个实用优点,包括例如更临床可行的周转时间(大约2周)、标准化信息学流程以及更易管理的成本。这种方法具有优于传统标记物诸如通过组织化学检测到的蛋白质表达的其他优点,因为它产生客观的(例如,突变负荷)而不是主观的量度(病理评分)(hansen和siu.jamaoncol2(1):15-6,2016)。此外,这个平台有助于同时检测与靶向疗法相关的可操作改变。
因此,本发明至少部分地提供了评估样品中的突变负荷的方法,所述方法通过以下方式来实现:从样品中提供亚基因组间隔集合的序列;并且确定突变负荷的值,其中值是亚基因组间隔集合中改变的数量的函数。在某些实施方案中,亚基因组间隔集合来自预定的基因集合,例如,不包括整个基因组或外显子组的预定的基因集合。在某些实施方案中,亚基因组间隔集合是编码亚基因组间隔集合。在其他实施方案中,亚基因组间隔集合包含编码亚基因组间隔和非编码亚基因组间隔。在某些实施方案中,突变负荷的值是亚基因组间隔集合中改变(例如,体细胞改变)的数量的函数。在某些实施方案中,改变的数量排除功能改变、种系改变或两者。在一些实施方案中,样品是肿瘤样品或源自肿瘤的样品。本文描述的方法还可包括例如以下的一种或多种:从样品中获取包括多个肿瘤成员的文库;通过杂交使文库与诱饵集合接触以提供选择的肿瘤成员,从而提供文库捕获;从文库捕获中获取来自肿瘤成员的包含改变的亚基因组间隔的读段;通过比对方法来比对读段;将来自读段的核苷酸值分配给预选的核苷酸位置;并且从分配的核苷酸位置集合中选择亚基因组间隔集合,其中亚基因组间隔集合来自预定的基因集合。还公开了用于评估样品中的突变负荷的系统。
首先定义某些术语。另外的术语定义在整个说明书中。
如本文所用,冠词“一个/种(a/an)”是指一个或多于一个(例如,至少一个)物品的语法对象。
“约”和“近似”通常意指在给定测量的性质或精度的情况下测量的量的可接受的误差程度。示例性误差程度在给定值或值范围的20%(%)内,通常在10%内,并且更通常,在5%内。
本文使用的术语“获取(acquire)”或“获取(acquiring)”是指通过“直接获取”或“间接获取”物理实体或值来获得物理实体或值(例如数值)的占有权。“直接获取”意味着执行一种方法(例如,执行合成或分析方法)以获得物理实体或值。“间接获取”是指从另一方或来源(例如,直接获取物理实体或值的第三方实验室)接收物理实体或值。直接获取物理实体包括执行包括物理物质例如起始材料的物理变化的方法。示例性变化包括由两种或更多种起始材料制备物理实体、剪切或片段化物质、分离或纯化物质、将两种或更多种分开的实体组合成混合物、执行包括破坏或形成共价或非共价键的化学反应。直接获取值包括执行包括样品或另一种物质的物理变化的方法,例如执行包括物质(例如,样品、分析物或试剂)的物理变化的分析方法(在本文中有时称为“物理分析”),执行分析方法,例如包括以下的一种或多种的方法:从另一种物质中分离或纯化一种物质,例如分析物或其片段或其他衍生物;将分析物或其片段或其他衍生物与另一种物质(例如,缓冲剂、溶剂或反应物)组合;或改变分析物或其片段或其他衍生物的结构,例如通过破坏或形成分析物的第一原子与第二原子之间的共价或非共价键;或通过改变试剂或其片段或其他衍生物的结构,例如通过破坏或形成试剂的第一原子与第二原子之间的共价或非共价键。
本文使用的术语“获取序列”或“获取读段”是指通过“直接获得”或“间接获得”序列或读段来获得核苷酸序列或氨基酸序列的占有权。“直接获取”序列或读段意味着执行一种方法(例如,执行合成或分析方法)以获得序列,诸如执行测序方法(例如,下一代测序(ngs)方法)。“间接获取”序列或读段是指从另一方或来源(例如,直接获取序列的第三方实验室)接收序列的信息或知识,或接收序列。获取的序列或读段不必是完整的序列,例如对至少一个核苷酸的测序或获得将本文公开的一个或多个改变鉴定为存在于受试者中的信息或知识,构成了获取序列。
直接获取序列或读段包括执行包括物理物质的物理变化的方法,所述物理物质例如起始材料,诸如组织或细胞样品,例如活组织检查物或分离的核酸(例如,dna或rna)样品。示例性变化包括由两种或更多种起始材料制备物理实体;剪切或片段化物质,诸如基因组dna片段;分离或纯化物质(例如,从组织中分离核酸样品);将两种或更多种分开的实体组合成混合物;执行包括破坏或形成共价或非共价键的化学反应。直接获取值包括执行包括如上所述的样品或另一种物质的物理变化的方法。
本文使用的术语“获取样品”是指通过“直接获取”或“间接获取”样品来获得样品,例如组织样品或核酸样品的占有权。“直接获取样品”意味着执行一种方法(例如,执行物理方法诸如手术或提取)以获得样品。“间接获取样品”是指从另一方或来源(例如,直接获取样品的第三方实验室)接收样品。直接获取样品包括执行包括物理物质的物理变化的方法,所述物理物质例如起始材料,诸如组织,例如人患者中的组织或先前已从患者中分离的组织。示例性变化包括从起始材料中制备物理实体,解剖或刮削组织;分离或纯化物质(例如,样品组织或核酸样品);将两种或更多种分开的实体组合成混合物;执行包括破坏或形成共价或非共价键的化学反应。直接获取样品包括执行包括例如以上所述的样品或另一种物质的物理变化的方法。
如本文所用的“比对选择因子”是指允许或引导比对方法(例如可优化预选亚基因组间隔的测序的比对算法或参数)选择的参数。比对选择因子可特定于以下的一种或多种或作为例如以下的一种或多种的函数而选择:
1.序列上下文,例如与所述亚基因组间隔的读段的错位倾向相关的亚基因组间隔(例如,待评估的预选核苷酸位置)的序列上下文。例如,在待评估的亚基因组间隔中或附近存有在基因组的其他地方重复的序列元件可导致错位,从而降低性能。可通过选择最小化错位的算法或算法参数来增强性能。在这种情况下,比对选择因子的值可以是序列上下文的函数,所述序列上下文例如存在或不存在预选长度的序列,所述预选长度的序列在基因组中(或在被分析的基因组的部分中)重复至少预选次数。
2.被分析的肿瘤类型。例如,特定的肿瘤类型可通过增加的缺失率来表征。因此,可通过选择对插入缺失更灵敏的算法或算法参数来增强性能。在这种情况下,比对选择因子的值可以是肿瘤类型的函数,例如肿瘤类型的标识符。在一个实施方案中,值是肿瘤类型例如恶性(恶化前)血液病的标识。
3.被分析的基因或基因类型,例如可被分析的基因或基因类型。举例来说,癌基因通常通过取代或框内插入缺失来表征。因此,可通过选择对这些变体特别灵敏并且对其他变体有特异性的算法或算法参数来增强性能。肿瘤抑制因子通常通过框移插入缺失来表征。因此,可通过选择对这些变体特别灵敏的算法或算法参数来增强性能。因此,可通过选择与亚基因组间隔匹配的算法或算法参数来增强性能。在这种情况下,比对选择因子的值可以是基因或基因类型的函数,例如基因或基因类型的标识符。在一个实施方案中,值是基因的标识。
4.被分析的位点(例如,核苷酸位置)。在这种情况下,比对选择因子的值可以是位点或位点类型的函数,例如位点或位点类型的标识符。在一个实施方案中,值是位点的标识。(例如,如果含有所述位点的基因与另一个基因高度同源,则正常/快速的短读取比对算法(例如,bwa)可能难以在两个基因之间进行区分,潜在地需要更密集的比对方法(smith-waterman)或甚至组装(arachne)。类似地,如果基因序列包含低复杂区域(例如,aaaaaa),则可能需要更密集的比对方法。
5.与被评估的亚基因组间隔相关的变体或变体类型。例如,取代、插入、缺失、易位或其他重排。因此,可通过选择对特定变体类型更灵敏的算法或算法参数来增强性能。在这种情况下,比对选择因子的值可以是变体类型的函数,例如变体类型的标识符。在一个实施方案中,值是变体类型例如取代的标识。
6.样品(ffpe或其他固定样品)的类型。样品类型/质量可影响误差(非参考序列的虚假观察)率。因此,可通过选择精确模拟样品中真实误差率的算法或算法参数来增强性能。在这种情况下,比对选择因子的值可以是样品类型的函数,例如样品类型的标识符。在一个实施方案中,值是样品类型(例如,固定样品)的标识。
本文所用的基因或基因产物(例如,标记基因或基因产物)的“改变”或“改变的结构”是指在基因或基因产物内存在一个突变或多个突变,例如与正常或野生型基因相比,影响基因或基因产物的完整性、序列、结构、量或活性的突变。与其在正常或健康组织或细胞(例如,对照)中的量、结构和/或活性相比,改变可以是癌症组织或癌细胞中的量、结构和/或活性的改变,并且与疾病状态诸如癌症相关。例如,与正常、健康的组织或细胞相比,与癌症相关或预测对抗癌治疗剂的响应性的改变可在癌症组织或癌细胞中具有改变的核苷酸序列(例如,突变)、氨基酸序列、染色体易位、染色体内倒位、拷贝数、表达水平、蛋白质水平、蛋白质活性、表观遗传修饰(例如,甲基化或乙酰化状态)或翻译后修饰。示例性突变包括但不限于点突变(例如,沉默、错义或无义)、缺失、插入、倒位、重复、扩增、易位、染色体间重排和染色体内重排。突变可存在于基因的编码区或非编码区中。在某些实施方案中,改变被检测为重排,例如包含一个或多个内含子或其片段的基因组重排(例如,5'-utr和/或3'-utr中的一个或多个重排)。在某些实施方案中,改变与表型相关(或无关),例如癌症表型(例如,癌症风险、癌症进展、癌症治疗或对癌症治疗的抗性中的一种或多种)。在一个实施方案中,改变与以下的一个或多个相关:癌症的遗传风险因素、阳性治疗响应预测因子、阴性治疗响应预测因子、阳性预后因素、阴性预后因素或诊断因素。
如本文所用,术语“插入缺失”是指在细胞核酸中的一个或多个核苷酸的插入、缺失或两者。在某些实施方案中,插入缺失包括一个或多个核苷酸的插入和缺失,其中插入和缺失都在核酸附近。在某些实施方案中,插入缺失导致核苷酸总数的净变化。在某些实施方案中,插入缺失导致约1至约50个核苷酸的净变化。
本文使用的术语“克隆谱”是指受试者间隔(或包含所述受试者间隔的细胞)的一个或多个序列(例如,等位基因或签名(signature))的发生、标识、变异性、分布、表达(亚基因组签名的转录拷贝的发生或水平)或丰度(例如,相对丰度)。在一个实施方案中,当所述受试者间隔的多个序列、等位基因或签名存在于样品中时,克隆谱是针对受试者间隔(或包含所述受试者间隔的细胞)的一个序列、等位基因或签名的相对丰度的值。例如,在一个实施方案中,克隆谱包括针对受试者间隔的多个vdj或vj组合中的一种或多种的相对丰度的值。在一个实施方案中,克隆谱包括针对受试者间隔的选择的v区段的相对丰度的值。在一个实施方案中,克隆谱包括受试者间隔的序列内的例如由体细胞高变引起时的多样性的值。在一个实施方案中,克隆谱包括序列、等位基因或签名表达的发生或水平的值,例如通过包含序列、等位基因或签名的表达的亚基因组间隔的发生或水平所证明。
本文使用的术语“表达的亚基因组间隔”是指亚基因组间隔的转录序列。在一个实施方案中,表达的亚基因组间隔的序列将不同于其转录自其中的亚基因组间隔,例如因为某个序列可能未被转录。
本文使用的术语“签名”是指受试者间隔的序列。签名可诊断受试者间隔处的多种可能性中的一种的发生,例如签名可诊断:在重排的重链或轻链可变区基因中选择的v区段的发生;选择的vj连接点的发生,例如在重排的重链可变区基因中选择的v区段和选择的j区段的发生。在一个实施方案中,签名包含多个特异性核酸序列。因此,签名不限于特异性核酸序列,而是足够独特以至于它可在受试者间隔处的第一组序列或可能性与受试者间隔处的第二组可能性之间进行区分,例如它可在第一v区段与第二v区段之间进行区分,例如允许评估各种v区段的用法。术语签名包括术语特异性签名,所述特异性签名是特异性核酸序列。在一个实施方案中,签名表示特定事件或是特定事件的产物,所述特定事件例如重排事件。
本文使用的术语“亚基因组间隔”是指基因组序列的一部分。在一个实施方案中,亚基因组间隔可以是单核苷酸位置,例如其核苷酸位置变体与肿瘤表型(正或负)相关。在一个实施方案中,亚基因组间隔包括多于一个核苷酸位置。此类实施方案包括长度为至少2、5、10、50、100、150或250个核苷酸位置的序列。亚基因组间隔可包含整个基因或其预选部分,例如编码区(或其部分)、预选的内含子(或其部分)或外显子(或其部分)。亚基因组间隔可包含天然存在的例如基因组dna、核酸的片段的全部或一部分。例如,亚基因组间隔可对应于受到测序响应的基因组dna的片段。在实施方案中,亚基因组间隔是来自基因组来源的连续序列。在实施方案中,亚基因组间隔包含在基因组中不连续的序列,例如它可包括在cdna的外显子-外显子连接处形成的连接点。
在一个实施方案中,亚基因组间隔对应于重排序列,例如b细胞或t细胞中的由于v区段与d区段、d区段与j区段、v区段与j区段、或j区段与一个类别区段的连接而产生的序列。
在一个实施方案中,亚基因组间隔没有多样性。
在一个实施方案中,亚基因组间隔存在多样性,例如亚基因组间隔由多于一个序列表示,例如覆盖vd序列的亚基因组间隔可由多于一个签名表示。
在一个实施方案中,亚基因组间隔包含或由以下组成:单核苷酸位置;基因内区或基因间区;外显子或内含子、其片段,通常外显子序列或其片段;编码区或非编码区,例如启动子、增强子、5’非翻译区(5’utr)、或3’非翻译区(3’utr)、或其片段;cdna或其片段;snp;体细胞突变、种系突变或两者;改变,例如点突变或单突变;缺失突变(例如,框内缺失、基因内缺失、全基因缺失);插入突变(例如,基因内插入);倒位突变(例如,染色体内倒位);连接突变;连接的插入突变;倒位的重复突变;串联重复(例如,染色体内串联重复);易位(例如,染色体易位、非相互易位);重排(例如,基因组重排(例如,一个或多个内含子或其片段的重排;重排内含子可包含5’-utr和/或3’-utr));基因拷贝数变化;基因表达变化;rna水平变化;或其组合。“基因的拷贝数”是指编码特定基因产物的细胞中的dna序列的数量。通常,针对给定的基因,哺乳动物的每个基因具有两个拷贝。可例如通过基因扩增或重复来增加拷贝数,或通过缺失来减少拷贝数。
本文使用的术语“受试者间隔”是指亚基因组间隔或表达的亚基因组间隔。在一个实施方案中,亚基因组间隔和表达的亚基因组间隔对应,意味着表达的亚基因组间隔包含从对应的亚基因组间隔表达的序列。在一个实施方案中,亚基因组间隔和表达的亚基因组间隔是非对应的,意味着表达的亚基因组间隔不包含从非对应的亚基因组间隔表达的序列,而是对应于不同的亚基因组间隔。在一个实施方案中,亚基因组间隔和表达的亚基因组间隔部分对应,意味着表达的亚基因组间隔包含从对应的亚基因组间隔表达的序列和从不同的对应亚基因组间隔表达的序列。
如本文所用,术语“文库”是指成员的集合。在一个实施方案中,文库包括核酸成员的集合,例如全基因组、亚基因组片段、cdna、cdna片段、rna(例如,mrna)、rna片段或其组合的集合。在一个实施方案中,文库成员中的一部分或全部包含衔接子序列。衔接子序列可位于一端或两端处。衔接子序列可例如用于测序方法(例如,ngs方法)、扩增、逆转录或克隆到载体中。
文库可以包括成员,例如靶成员(例如,肿瘤成员、参考成员、pgx成员或其组合)的集合。文库的成员可来自单个个体。在实施方案中,文库可包括来自多于一个受试者(例如,2、3、4、5、6、7、8、9、10、20、30或更多个受试者)的成员,例如来自不同受试者的两个或更多个文库可组合以形成包括来自多于一个受试者的成员的文库。在一个实施方案中,受试者是患有癌症或肿瘤或处于患有癌症或肿瘤风险的人。
“文库捕获”是指文库的子集,例如富含预选亚基因组间隔的子集,例如通过与预选诱饵杂交捕获的产物。
如本文所用,“成员”或“文库成员”或其他类似术语是指作为文库成员的核酸分子,例如dna、rna或其组合。通常,成员是dna分子,例如基因组dna或cdna。成员可以是片段化的(例如,剪切的)或酶促制备的基因组dna。成员包含来自受试者的序列,并且还可包含并非源自受试者的序列,例如衔接子序列、引物序列或其他允许标识的序列,例如“条形码”序列。
如本文所用,“诱饵”是杂交捕获试剂的类型。诱饵可以是核酸分子,例如dna或rna分子,其可与靶核酸杂交(例如,互补),从而允许捕获靶核酸。在一个实施方案中,诱饵是rna分子(例如,天然存在的或修饰的rna分子);dna分子(例如,天然存在的或修饰的dna分子)、或其组合。在其他实施方案中,诱饵包含结合实体,例如亲和标签,所述诱饵例如通过与结合实体结合来允许捕获和分离杂交体,所述杂交体由诱饵和与诱饵杂交的核酸形成。在一个实施方案中,诱饵适合于液相杂交。在一个实施方案中,诱饵是双环核酸(bna)分子。
如本文所用,“诱饵集合”是指一个或多个诱饵分子。
“结合实体”意指可直接或间接连接分子标签的任何分子,所述结合实体能够特异性结合分析物。结合实体可以是每个诱饵序列上的亲和标签。在某些实施方案中,结合实体通过结合配偶体(诸如亲和素分子)或结合半抗原或其抗原结合片段的抗体来允许从杂交混合物中分离诱饵/成员杂交体。示例性结合实体包括但不限于生物素分子、半抗原、抗体、抗体结合片段、肽和蛋白质。
“互补”是指两条核酸链的区域之间或相同核酸链的两个区域之间的序列互补性。已知第一核酸区域的腺嘌呤残基能够与第二核酸区域的残基形成特定的氢键(“碱基配对”),如果残基是胸腺嘧啶或尿嘧啶,则所述第二核酸区域与第一区域反平行。类似地,已知第一核酸链的胞嘧啶残基能够与第二核酸链的残基碱基配对,如果残基是鸟嘌呤,则所述第二核酸链与第一链反平行。如果当两个区域以反平行方式布置,第一区域的至少一个核苷酸残基能够与第二区域的残基碱基配对,则核酸的第一区域与相同或不同核酸的第二区域互补。在某些实施方案中,第一区域包含第一部分,并且第二区域包含第二部分,由此,当第一部分和第二部分以反平行方式布置时,第一部分的至少约50%、至少约75%、至少约90%或至少约95%的核苷酸残基能够与第二部分中的核苷酸残基碱基配对。在其他实施方案中,第一部分的所有核苷酸残基能够与第二部分中的核苷酸残基碱基配对。
术语“癌症”或“肿瘤”在本文中可互换使用。这些术语是指存在具有致癌细胞典型特征的细胞,诸如不受控制的增殖、永生、转移潜能、快速的生长和增殖率以及某些特征性形态特征。癌细胞通常呈肿瘤的形式,但是此类细胞可单独存在于动物体内,或者可以是无致瘤性癌细胞,诸如白血病细胞。这些术语包括实体瘤、软组织肿瘤或转移病变。如本文所用,术语“癌症”包括恶化前期癌症以及恶性癌症。
如本文所用的“可能”或“增加的可能性”是指物品、物体、事情或人将发生的概率增加。因此,在一个实例中,相对于参考受试者或受试者组,可能响应于治疗的受试者具有增加的响应于治疗的概率。
“不太可能”是指相对于参考,事件、物品、物体、事情或人将发生的概率降低。因此,相对于参考受试者或受试者组,不太可能响应于治疗的受试者具有降低的响应于治疗的概率。
“对照成员”是指具有来自非肿瘤细胞的序列的成员。
如本文所用,“插入缺失比对序列选择因子”是指在预选的插入缺失的情况下,允许或引导读段待与之比对的序列的选择的参数。使用这种序列可优化包含插入缺失的预选亚基因组间隔的测序。插入缺失比对序列选择因子的值是预选插入缺失的函数,例如插入缺失的标识符。在一个实施方案中,值是插入缺失的标识。
如本文所用,“下一代测序或ngs或ng测序”是指以高通量方式(例如,同时测序大于103、104、105或更多个分子)确定单个核酸分子(例如,在单分子测序中)或单个核酸分子的克隆扩增代用品的核苷酸序列的任何测序方法。在一个实施方案中,可通过计数由测序实验产生的数据中其同源序列的相对发生次数来估计文库中核酸种类的相对丰度。下一代测序方法是本领域中已知的,并且描述于例如metzker,m.(2010)naturebiotechnologyreviews11:31-46中,所述参考文献以引用的方式并入本文。下一代测序可检测存在于样品中小于5%的核酸中的变体。
如本文所提及的“核苷酸值”代表占据或分配给预选核苷酸位置的核苷酸的标识。典型的核苷酸值包括:缺少(例如,缺失的);附加(例如,一个或多个核苷酸的插入,其标识可以或可以不包括在内);或存在(占据);a;t;c;或g。其他值可以是例如非y,其中y是a、t、g或c;a或x,其中x是t、g或c中的一种或两种;t或x,其中x是a、g或c中的一种或两种;g或x,其中x是t、a或c中的一种或两种;c或x,其中x是t、g或a中的一种或两种;嘧啶核苷酸;或嘌呤核苷酸。核苷酸值可以是1个或更多个例如2、3或4个碱基(或本文描述的其他值,例如缺少或附加)在核苷酸位置处的频率。例如,核苷酸值可包括在核苷酸位置处的a的频率和g的频率。
除非上下文另有明确说明,否则“或”在本文中用于意指术语“和/或”并且可与术语“和/或”互换使用。除非上下文另有明确说明,否则在本文的某些地方使用术语“和/或”并不意味着使用术语“或”不能与术语“和/或”互换。
“基本对照(primarycontrol)”是指肿瘤样品中除nat组织之外的非肿瘤组织。血液是典型的基本对照。
如本文所用,“重排比对序列选择因子”是指在预选的重排的情况下,允许或引导读段待与之比对的序列的选择的参数。使用这种序列可优化包含重排的预选亚基因组间隔的测序。重排比对序列选择因子的值是预选重排的函数,例如重排的标识符。在一个实施方案中,值是重排的标识。“插入缺失比对序列选择因子”(在本文其他地方也定义)是重排比对序列选择因子的一个实例。
“样品”、“组织样品”、“患者样品”、“患者细胞或组织样品”或“样本”包含从受试者或患者中获得的组织、细胞,例如循环细胞。组织样品的来源可以是来自新鲜、冷冻和/或保存的器官、组织样品、活组织检查物或抽吸物的实体组织;血液或任何血液组分;体液,诸如脑脊髓液、羊水、腹膜液或间质液;或来自受试者妊娠或发育的任何时间的细胞。组织样品可包含本质上不与组织天然混合的化合物,诸如防腐剂、抗凝血剂、缓冲剂、固定剂、营养素、抗生素等。在一个实施方案中,将样品保存为冷冻的样品或甲醛固定的或多聚甲醛固定的石蜡包埋(ffpe)组织制剂。例如,样品可被包埋在基质中,例如ffpe块或冷冻样品。在另一个实施方案中,样品是血液样品。在另一个实施方案中,样品是骨髓抽吸物样品。在另一个实施方案中,样品包含循环肿瘤dna(ctdna)。在另一个实施方案中,样品包含循环肿瘤细胞(ctc)。
在一个实施方案中,样品是与肿瘤相关的细胞,例如肿瘤细胞或肿瘤浸润淋巴细胞(til)。在一个实施方案中,样品是肿瘤样品,例如包含一个或多个恶化前或恶性细胞。在一个实施方案中,样品从恶性(恶化前)血液病,例如本文描述的恶性(恶化前)血液病中获取。在某些实施方案中,样品例如肿瘤样品从实体瘤、软组织肿瘤或转移病变中获取。在其他实施方案中,样品例如肿瘤样品包含来自手术切缘的组织或细胞。在另一个实施方案中,样品例如肿瘤样品包含一个或多个循环肿瘤细胞(ctc)(例如,从血液样品中获取的ctc)。在一个实施方案中,样品是与肿瘤无关的细胞,例如非肿瘤细胞或外周血淋巴细胞。
如本文所用,“灵敏度”是方法检测异种序列群体中预选序列变体的能力的量度。如果给定其中预选序列变体作为样品中至少f%序列存在的样品,方法可在c%预选置信度、s%次数下检测到预选序列,则方法针对f%的变体的灵敏度为s%。举例来说,如果给定其中预选变体序列作为样品中至少5%序列存在的样品,方法可在99%预选置信度、10次中有9次的情况下检测到预选序列(f=5%;c=99%;s=90%),则方法针对5%的变体的灵敏度为90%。示例性灵敏度包括在c=90%、95%、99%和99.9%的置信度水平下,针对f=1%、5%、10%、20%、50%、100%下的序列变体的那些s=90%、95%、99%。
如本文所用,“特异性”是方法区分真正存在的预选序列变体与测序假象或其他密切相关序列的能力的量度。它是避免假阳性检测的能力。假阳性检测可由样品制备过程中引入到所关注的序列中的错误、测序错误或密切相关序列(像基因家族的假基因或成员)的无意测序引起。如果应用于n总序列的样品集合(其中x真正序列是真正的变体并且和x非真正是非真正的变体)时,方法将至少x%的非真正的变体选择为非变体,则方法的特异性为x%。例如,如果应用于1,000个序列的样品集合(其中500个序列是真正的变体并且500个是非真正的变体)时,方法将至少90%的500个非真正的变体序列选择为非变体,则方法的特异性为90%。示例性特异性包括90%、95%、98%和99%。
如本文所用的“肿瘤核酸样品”是指来自肿瘤或癌症样品的核酸分子。通常,它是dna,例如来自肿瘤或癌症样品的基因组dna或源自rna的cdna。在某些实施方案中,纯化或分离肿瘤核酸样品(例如,将它从其天然状态中移出)。
如本文所用的“对照”或“参考”“核酸样品”是指来自对照或参考样品的核酸分子。通常,它是dna,例如在基因或基因产物中不含有改变或变异的基因组dna或源自rna的cdna。在某些实施方案中,参考或对照核酸样品是野生型或未突变的序列。在某些实施方案中,纯化或分离参考核酸样品(例如,将它从其天然状态中移出)。在其他实施方案中,参考核酸样品来自非肿瘤样品,例如血液对照、正常相邻组织(nat)、或来自相同或不同受试者的任何其他非癌样品。
“测序”核酸分子需要确定分子(例如,dna分子、rna分子、或源自rna分子的cdna分子)中至少1个核苷酸的标识。在实施方案中,确定分子中少于所有核苷酸的标识。在其他实施方案中,确定分子中大部分或全部核苷酸的标识。
如本文所用,“阈值”是作为读段数量的函数的值,需要存在所述读段以将核苷酸值分配给受试者间隔(例如,亚基因组间隔或表达的亚基因组间隔)。例如,它是在核苷酸位置处具有特定核苷酸值例如“a”的读段数量的函数,需要所述读段以将所述核苷酸值分配给亚基因组间隔中的所述核苷酸位置。阈值可例如表示为读段数量例如整数,或具有预选值的读段比例(或其函数)。举例来说,如果阈值是x,并且存在核苷酸值为“a”的x+1个读段,则将“a”的值分配给受试者间隔(例如,亚基因组间隔或表达的亚基因组间隔)中的预选位置。阈值还可表示为突变或变体期望值(突变频率)或贝叶斯先验值的函数。在一个实施方案中,预选突变频率将需要具有核苷酸值(例如,预选位置处的a或g)的预选数量或比例的读段以识别所述核苷酸值。在实施方案中,阈值可以是突变期望值(例如突变频率)和肿瘤类型的函数。例如,如果患者具有第一肿瘤类型,则预选核苷酸位置处的预选变体可具有第一阈值,如果患者具有第二肿瘤类型,则预选核苷酸位置处的预选变体可具有第二阈值。
如本文所用,“靶成员”是指人们希望从核酸文库中分离的核酸分子。在一个实施方案中,靶成员可以是如本文所述的肿瘤成员、参考成员、对照成员或pgx成员。
如本文所用,“肿瘤成员”或其他类似术语(例如,“肿瘤或癌症相关成员”)是指具有来自肿瘤细胞的序列的成员。在一个实施方案中,肿瘤成员包含具有序列(例如,核苷酸序列)的受试者间隔(例如,亚基因组间隔或表达的亚基因组间隔),所述序列具有与癌症表型相关的改变(例如,突变)。在其他实施方案中,肿瘤成员包含具有野生型序列(例如,野生型核苷酸序列)的受试者间隔(例如,亚基因组间隔或表达的亚基因组间隔)。例如,来自杂合或纯合野生型等位基因的受试者间隔(例如,亚基因组间隔或表达的亚基因组间隔)存在于癌细胞中。肿瘤成员可包括参考成员或pgx成员。
如本文所用,“参考成员”或其他类似术语(例如,“对照成员”)是指包含受试者间隔(例如,亚基因组间隔或表达的亚基因组间隔)的成员,所述受试者间隔具有与癌症表型无关的序列(例如,核苷酸序列)。在一个实施方案中,参考成员包含突变与癌症表型相关时的基因或基因产物的野生型或未突变的核苷酸序列。参考成员可存在于癌细胞或非癌症细胞中。
如本文所用,“pgx成员”或其他类似术语是指包含受试者间隔(例如,亚基因组间隔或表达的亚基因组间隔)的成员,所述受试者间隔与基因的药物遗传学或药物基因组学谱相关。在一个实施方案中,pgx成员包含snp(例如,本文所述的snp)。在其他实施方案中,pgx成员包含根据表1-4或图3a-4d的受试者间隔(例如,亚基因组间隔或表达的亚基因组间隔)。
如本文所用,“变体”是指可存在于可具有多于一种结构的亚基因组间隔处的结构,例如多态基因座处的等位基因。
如本文所用,“x是y的函数”意味着例如一个变量x与另一个变量y相关。在一个实施方案中,如果x是y的函数,则可暗示x与y之间的因果关系,但不一定存在。
标题例如(a)、(b)、(i)等仅仅是为了便于阅读说明书和权利要求书而提供。在说明书或权利要求书中使用标题不需要按字母或数字顺序或其中提供它们的顺序来执行步骤或要素。
突变负荷
如本文所用,术语“突变负荷(mutationload)”或“突变负荷(mutationalload)”是指在预定的基因集合中(例如,预定的基因集合的编码区中)每个预选单位(例如,每兆碱基)的改变(例如一个或多个改变,例如一个或多个体细胞改变)的水平例如数量。可例如基于全基因组或外显子组,或基于基因组或外显子组的子集来测量突变负荷。在某些实施方案中,可对基于基因组或外显子组的子集测量的突变负荷进行外推,以确定全基因组或外显子组突变负荷。
在某些实施方案中,在来自受试者(例如本文描述的受试者)的样品中测量突变负荷,所述样品例如肿瘤样品(例如,肿瘤样品或源自肿瘤的样品)。在某些实施方案中,突变负荷表示为例如来自参考群体的样品中的突变负荷之中的百分位数。在某些实施方案中,参考群体包括与受试者患有相同类型癌症的患者。在其他实施方案中,参考群体包括正在接受或已接受与受试者相同类型疗法的患者。在某些实施方案中,通过本文描述的方法,例如通过评估表1-4或图3a-4d中所示的预定的基因集合中的改变(例如,体细胞改变)的水平而获得的突变负荷,与全基因组或外显子组突变负荷相关。
术语“突变负荷”、“突变负荷”、“突变负荷(mutationburden)”和“突变负荷(mutationalburden)”在本文中可互换使用。在肿瘤的背景下,突变负荷在本文中又称为“肿瘤突变负荷”、“肿瘤突变负荷”或“tmb”。
基因选择
选择的基因或基因产物(在本文中又称为“靶基因或基因产物”)可包含受试者间隔(例如,亚基因组间隔、表达的亚基因组间隔、或两者),所述受试者间隔包含基因内区或基因间区。例如,受试者间隔(例如,亚基因组间隔或表达的亚基因组间隔)可包含外显子或内含子或其片段,通常外显子序列或其片段。受试者间隔(例如,亚基因组间隔或表达的亚基因组间隔)可包含编码区或非编码区,例如启动子、增强子、5’非翻译区(5’utr)、或3’非翻译区(3’utr)、或其片段。在其他实施方案中,受试者间隔包含cdna或其片段。在其他实施方案中,受试者间隔包含例如本文所述的snp。
在其他实施方案中,受试者间隔(亚基因组间隔、表达的亚基因组间隔、或两者)包含基因组(例如,本文所述的一个或多个受试者间隔(例如,亚基因组间隔、表达的亚基因组间隔、或两者))中的基本上所有的外显子(例如,来自选择的所关注的基因或基因产物(例如,与本文所述的癌症表型相关的基因或基因产物)的外显子)。在一个实施方案中,受试者间隔(例如,亚基因组间隔或表达的亚基因组间隔)包含体细胞突变、种系突变或两者。在一个实施方案中,受试者间隔(例如,亚基因组间隔或表达的亚基因组间隔)包含改变,例如点突变或单突变、缺失突变(例如,框内缺失、基因内缺失、全基因缺失)、插入突变(例如,基因内插入)、倒位突变(例如,染色体内倒位)、连接突变、连接的插入突变、倒位的重复突变、串联重复(例如,染色体内串联重复)、易位(例如,染色体易位、非相互易位)、重排、基因拷贝数变化、或其组合。在某些实施方案中,受试者间隔(例如,亚基因组间隔或表达的亚基因组间隔)构成样品中肿瘤细胞基因组编码区的小于5%、1%、0.5%、0.1%、0.01%、0.001%。在其他实施方案中,受试者间隔(例如,亚基因组间隔、表达的亚基因组间隔、或两者)不涉及疾病,例如与本文所述的癌症表型无关。
在一个实施方案中,靶基因或基因产物是生物标记物。如本文所用,“生物标记物”或“标记物”是可改变的基因、mrna或蛋白质,其中所述改变与癌症相关。与其在正常或健康组织或细胞(例如,对照)中的量、结构和/或活性相比,改变可以是癌症组织或癌细胞中的量、结构和/或活性的改变,并且与疾病状态诸如癌症相关。例如,与正常、健康的组织或细胞相比,与癌症相关或预测对抗癌治疗剂的响应性的标记物可在癌症组织或癌细胞中具有改变的核苷酸序列、氨基酸序列、染色体易位、染色体内倒位、拷贝数、表达水平、蛋白质水平、蛋白质活性、表观遗传修饰(例如,甲基化或乙酰化状态)或翻译后修饰。此外,“标记物”包括当存在于与疾病状态诸如癌症相关的组织或细胞中时,其结构例如通过取代、缺失或插入而改变例如突变(包含突变),例如在核苷酸或氨基酸水平下与野生型序列不同的分子。
在一个实施方案中,靶基因或基因产物包含单核苷酸多态性(snp)。在另一个实施方案中,基因或基因产物具有小的缺失,例如小的基因内缺失(例如,框内或框移缺失)。在另一个实施方案中,靶序列由整个基因的缺失产生。在另一个实施方案中,靶序列具有小的插入,例如小的基因内插入。在一个实施方案中,靶序列由倒位例如染色体内倒位产生。在另一个实施方案中,靶序列由染色体间易位产生。在另一个实施方案中,靶序列具有串联重复。在一个实施方案中,靶序列具有不期望的特征(例如,高gc含量或重复元件)。在另一个实施方案中,靶序列具有例如由于其重复性,本身不能被成功靶向的一部分核苷酸序列。在一个实施方案中,靶序列由可变剪接产生。在另一个实施方案中,靶序列选自根据表1-4或图3a-4d的基因或基因产物、或其片段。
在一个实施方案中,靶基因或基因产物或其片段是抗体基因或基因产物、免疫球蛋白超家族受体(例如,b细胞受体(bcr)或t细胞受体(tcr))基因或基因产物、或其片段。
人抗体分子(和b细胞受体)由重链和轻链组成,其中恒定(c)区和可变(v)区由至少以下三个基因座上的基因编码。
1.14号染色体上的免疫球蛋白重链基因座(igh@),其含有免疫球蛋白重链的基因区段;
2.2号染色体上的免疫球蛋白κ(κ)基因座(igk@),其含有免疫球蛋白轻链的基因区段;
3.22号染色体上的免疫球蛋白λ(λ)基因座(igl@),其含有免疫球蛋白轻链的基因区段。
每个重链和轻链基因包含抗体蛋白可变区的三种不同类型基因区段的多个拷贝。例如,免疫球蛋白重链区可包含五种不同类别γ、δ、α、μ和ε、44个可变(v)基因区段、27个多样性(d)基因区段和6个连接(j)基因区段中的一种。轻链还可以具有数个v和j基因区段,但不具有d基因区段。λ轻链具有7个可能的c区,并且κ轻链具有1个。
免疫球蛋白重链基因座(igh@)是人14号染色体上的区域,所述区域包含人抗体(或免疫球蛋白)重链的基因。例如,igh基因座包括ighv(可变)、ighd(多样性)、ighj(连接)和ighc(恒定)基因。编码免疫球蛋白重链的示例性基因包括但不限于ighv1-2、ighv1-3、ighv1-8、ighv1-12、ighv1-14、ighv1-17、ighv1-18、ighv1-24、ighv1-45、ighv1-46、ighv1-58、ighv1-67、ighv1-68、ighv1-69、ighv1-38-4、ighv1-69-2、ighv2-5、ighv2-10、ighv2-26、ighv2-70、ighv3-6、ighv3-7、ighv3-9、ighv3-11、ighv3-13、ighv3-15、ighv3-16、ighv3-19、ighv3-20、ighv3-21、ighv3-22、ighv3-23、ighv3-25、ighv3-29、ighv3-30、ighv3-30-2、ighv3-30-3、ighv3-30-5、ighv3-32、ighv3-33、ighv3-33-2、ighv3-35、ighv3-36、ighv3-37、ighv3-38、ighv3-41、ighv3-42、ighv3-43、ighv3-47、ighv3-48、ighv3-49、ighv3-50、ighv3-52、ighv3-53、ighv3-54、ighv3-57、ighv3-60、ighv3-62、ighv3-63、ighv3-64、ighv3-65、ighv3-66、ighv3-71、ighv3-72、ighv3-73、ighv3-74、ighv3-75、ighv3-76、ighv3-79、ighv3-38-3、ighv3-69-1、ighv4-4、ighv4-28、ighv4-30-1、ighv4-30-2、ighv4-30-4、ighv4-31、ighv4-34、ighv4-39、ighv4-55、ighv4-59、ighv4-61、ighv4-80、ighv4-38-2、ighv5-51、ighv5-78、ighv5-10-1、ighv6-1、ighv7-4-1、ighv7-27、ighv7-34-1、ighv7-40、ighv7-56、ighv7-81、ighvii-1-1、ighvii-15-1、ighvii-20-1、ighvii-22-1、ighvii-26-2、ighvii-28-1、ighvii-30-1、ighvii-31-1、ighvii-33-1、ighvii-40-1、ighvii-43-1、ighvii-44-2、ighvii-46-1、ighvii-49-1、ighvii-51-2、ighvii-53-1、ighvii-60-1、ighvii-62-1、ighvii-65-1、ighvii-67-1、ighvii-74-1、ighvii-78-1、ighviii-2-1、ighviii-5-1、ighviii-5-2、ighviii-11-1、ighviii-13-1、ighviii-16-1、ighviii-22-2、ighviii-25-1、ighviii-26-1、ighviii-38-1、ighviii-44、ighviii-47-1、ighviii-51-1、ighviii-67-2、ighviii-67-3、ighviii-67-4、ighviii-76-1、ighviii-82、ighviv-44-1、ighd1-1、ighd1-7、ighd1-14、ighd1-20、ighd1-26、ighd2-2、ighd2-8、ighd2-15、ighd2-21、ighd3-3、ighd3-9、ighd3-10、ighd3-16、ighd3-22、ighd4-4、ighd4-11、ighd4-17、ighd4-23、ighd5-5、ighd5-12、ighd5-18、ighd5-24、ighd6-6、ighd6-13、ighd6-19、ighd6-25、ighd7-27、ighj1、ighj1p、ighj2、ighj2p、ighj3、ighj3p、ighj4、ighj5、ighj6、igha1、igha2、ighg1、ighg2、ighg3、ighg4、ighgp、ighd、ighe、ighep1、ighm和ighv1-69d。
免疫球蛋白κ基因座(igk@)是人2号染色体上的区域,所述区域包含抗体(或免疫球蛋白)的κ(κ)轻链的基因。例如,igk基因座包括igkv(可变)、igkj(连接)和igkc(恒定)基因。编码免疫球蛋白κ轻链的示例性基因包括但不限于igkv1-5、igkv1-6、igkv1-8、igkv1-9、igkv1-12、igkv1-13、igkv1-16、igkv1-17、igkv1-22、igkv1-27、igkv1-32、igkv1-33、igkv1-35、igkv1-37、igkv1-39、igkv1d-8、igkv1d-12、igkv1d-13、igkv1d-16igkv1d-17、igkv1d-22、igkv1d-27、igkv1d-32、igkv1d-33、igkv1d-35、igkv1d-37、igkv1d-39、igkv1d-42、igkv1d-43、igkv2-4、igkv2-10、igkv2-14、igkv2-18、igkv2-19、igkv2-23、igkv2-24、igkv2-26、igkv2-28、igkv2-29、igkv2-30、igkv2-36、igkv2-38、igkv2-40、igkv2d-10、igkv2d-14、igkv2d-18、igkv2d-19、igkv2d-23、igkv2d-24、igkv2d-26、igkv2d-28、igkv2d-29、igkv2d-30、igkv2d-36、igkv2d-38、igkv2d-40、igkv3-7、igkv3-11、igkv3-15、igkv3-20、igkv3-25、igkv3-31、igkv3-34、igkv3d-7、igkv3d-11、igkv3d-15、igkv3d-20、igkv3d-25、igkv3d-31、igkv3d-34、igkv4-1、igkv5-2、igkv6-21、igkv6d-21、igkv6d-41、igkv7-3、igkj1、igkj2、igkj3、igkj4、igkj5和igkc。
免疫球蛋白λ基因座(igl@)是人22号染色体上的区域,所述区域包含抗体(或免疫球蛋白)的λ轻链的基因。例如,igl基因座包括iglv(可变)、iglj(连接)和iglc(恒定)基因。编码免疫球蛋白λ轻链的示例性基因包括但不限于iglv1-36、iglv1-40、iglv1-41、iglv1-44、iglv1-47、iglv1-50、iglv1-51、iglv1-62、iglv2-5、iglv2-8、iglv2-11、iglv2-14、iglv2-18、iglv2-23、iglv2-28、iglv2-33、iglv2-34、iglv3-1、iglv3-2、iglv3-4、iglv3-6、iglv3-7、iglv3-9、iglv3-10、iglv3-12、iglv3-13、iglv3-15、iglv3-16、iglv3-17、iglv3-19、iglv3-21、iglv3-22、iglv3-24、iglv3-25、iglv3-26、iglv3-27、iglv3-29、iglv3-30、iglv3-31、iglv3-32、iglv4-3、iglv4-60、iglv4-69、iglv5-37、iglv5-39、iglv5-45、iglv5-48、iglv5-52、iglv6-57、iglv7-35、iglv7-43、iglv7-46、iglv8-61、iglv9-49、iglv10-54、iglv10-67、iglv11-55、iglvi-20、iglvi-38、iglvi-42、iglvi-56、iglvi-63、iglvi-68、iglvi-70、iglviv-53、iglviv-59、iglviv-64、iglviv-65、iglviv-66-1、iglvv-58、iglvv-66、iglvvi-22-1、iglvvi-25-1、iglvvii-41-1、iglj1、iglj2、iglj3、iglj4、iglj5、iglj6、iglj7、iglc1、iglc2、iglc3、iglc4、iglc5、iglc6和iglc7。
b细胞受体(bcr)由两部分组成:i)一种同种型的膜结合免疫球蛋白分子(例如,igd或igm)。除了存在完整的膜结构域外,这些可与其分泌形式相同,和ii)信号转导部分:由二硫键结合在一起的称为ig-α/ig-β(cd79)的异源二聚体。二聚体的每个成员跨越质膜并且具有胞质尾区,所述胞质尾区带有免疫受体酪氨酸活化基序(itam)。
t细胞受体(tcr)由两种不同的蛋白质链组成(即,异源二聚体)。在95%的t细胞中,所述t细胞受体由α(α)和β(β)链组成,而在5%的t细胞中,所述t细胞受体由γ(γ)和δ(δ)链组成。这个比率可在个体发育和患病状态期间发生变化。t细胞受体基因与免疫球蛋白基因类似,因为它们的β和δ链中包含多个v、d和j基因区段(以及α和γ链中的v和j基因区段),所述基因区段在淋巴细胞发育以给每个细胞提供独特的抗原受体的过程中重排。
t细胞受体α基因座(tra)是人14号染色体上的区域,所述区域包含tcrα链的基因。例如,tra基因座包括例如trav(可变)、traj(连接)和trac(恒定)基因。编码t细胞受体α链的示例性基因包括但不限于trav1-1、trav1-2、trav2、trav3、trav4、trav5、trav6、trav7、trav8-1、trav8-2、trav8-3、trav8-4、trav8-5、trav8-6、trav8-7、trav9-1、trav9-2、trav10、trav11、trav12-1、trav12-2、trav12-3、trav13-1、trav13-2、trav14dv4、trav15、trav16、trav17、trav18、trav19、trav20、trav21、trav22、trav23dv6、trav24、trav25、trav26-1、trav26-2、trav27、trav28、trav29dv5、trav30、trav31、trav32、trav33、trav34、trav35、trav36dv7、trav37、trav38-1、trav38-2dv8、trav39、trav40、trav41、traj1、traj2、traj3、traj4、traj5、traj6、traj7、traj8、traj9、traj10、traj11、traj12、traj13、traj14、traj15、traj16、traj17、traj18、traj19、traj20、traj21、traj22、traj23、traj24、traj25、traj26、traj27、traj28、traj29、traj30、traj31、traj32、traj33、traj34、traj35、traj36、traj37、traj38、traj39、traj40、traj41、traj42、traj43、traj44、traj45、traj46、traj47、traj48、traj49、traj50、traj51、traj52、traj53、traj54、traj55、traj56、traj57、traj58、traj59、traj60、traj61和trac。
t细胞受体β基因座(trb)是人7号染色体上的区域,所述区域包含tcrβ链的基因。例如,trb基因座包括例如trbv(可变)、trbd(多样性)、trbj(连接)和trbc(恒定)基因。编码t细胞受体β链的示例性基因包括但不限于trbv1、trbv2、trbv3-1、trbv3-2、trbv4-1、trbv4-2、trbv4-3、trbv5-1、trbv5-2、trbv5-3、trbv5-4、trbv5-5、trbv5-6、trbv5-7、trbv6-2、trbv6-3、trbv6-4、trbv6-5、trbv6-6、trbv6-7、trbv6-8、trbv6-9、trbv7-1、trbv7-2、trbv7-3、trbv7-4、trbv7-5、trbv7-6、trbv7-7、trbv7-8、trbv7-9、trbv8-1、trbv8-2、trbv9、trbv10-1、trbv10-2、trbv10-3、trbv11-1、trbv11-2、trbv11-3、trbv12-1、trbv12-2、trbv12-3、trbv12-4、trbv12-5、trbv13、trbv14、trbv15、trbv16、trbv17、trbv18、trbv19、trbv20-1、trbv21-1、trbv22-1、trbv23-1、trbv24-1、trbv25-1、trbv26、trbv27、trbv28、trbv29-1、trbv30、trbva、trbvb、trbv5-8、trbv6-1、trbd1、trbd2、trbj1-1、trbj1-2、trbj1-3、trbj1-4、trbj1-5、trbj1-6、trbj2-1、trbj2-2、trbj2-2p、trbj2-3、trbj2-4、trbj2-5、trbj2-6、trbj2-7、trbc1和trbc2。
t细胞受体δ基因座(trd)是人14号染色体上的区域,所述区域包含tcrδ链的基因。例如,trd基因座包括例如trdv(可变)、trdj(连接)和trdc(恒定)基因。编码t细胞受体δ链的示例性基因包括但不限于trdv1、trdv2、trdv3、trdd1、trdd2、trdd3、trdj1、trdj2、trdj3、trdj4和trdc。
t细胞受体γ基因座(trg)是人7号染色体上的区域,所述区域包含tcrγ链的基因。例如,trg基因座包括例如trgv(可变)、trgj(连接)和trgc(恒定)基因。编码t细胞受体γ链的示例性基因包括但不限于trgv1、trgv2、trgv3、trgv4、trgv5、trgv5p、trgv6、trgv7、trgv8、trgv9、trgv10、trgv11、trgva、trgvb、trgj1、trgj2、trgjp、trgjp1、trgjp2、trgc1和trgc2。
示例性癌症包括但不限于b细胞癌,例如多发性骨髓瘤、黑色素瘤、乳腺癌、肺癌(诸如非小细胞肺癌或nsclc)、支气管癌、结直肠癌、前列腺癌、胰腺癌、胃癌、卵巢癌、膀胱癌、脑或中枢神经系统癌症、周围神经系统癌症、食道癌、宫颈癌、子宫或子宫内膜癌、口腔癌或咽癌、肝癌、肾癌、睾丸癌、胆道癌、小肠或阑尾癌、唾液腺癌、甲状腺癌、肾上腺癌、骨肉瘤、软骨肉瘤、血液组织癌、腺癌、炎性肌纤维母细胞瘤、胃肠道间质瘤(gist)、结肠癌、多发性骨髓瘤(mm)、骨髓增生异常综合征(mds)、骨髓增生性疾病(mpd)、急性淋巴细胞白血病(all)、急性髓细胞白血病(aml)、慢性髓细胞白血病(cml)、慢性淋巴细胞白血病(cll)、真性红细胞增多症、霍奇金淋巴瘤(hodgkinlymphoma)、非霍奇金淋巴瘤(nhl)、软组织肉瘤、纤维肉瘤、粘液肉瘤、脂肪肉瘤、成骨肉瘤、脊索瘤、血管肉瘤、内皮肉瘤、淋巴管肉瘤、淋巴管内皮瘤、滑膜瘤、间皮瘤、尤文氏瘤(ewing'stumor)、平滑肌肉瘤、横纹肌肉瘤、鳞状细胞癌、基底细胞癌、腺癌、汗腺癌、皮脂腺癌、乳头状癌、乳头状腺癌、髓样癌、支气管肺癌、肾细胞癌、肝癌、胆管癌、绒毛膜癌、精原细胞瘤、胚胎癌、维尔姆斯瘤、膀胱癌、上皮癌、胶质瘤、星形细胞瘤、成神经管细胞瘤、颅咽管瘤、室管膜瘤、松果体瘤、血管母细胞瘤、听神经瘤、少突神经胶质瘤、脑脊膜瘤、神经母细胞瘤、视网膜母细胞瘤、滤泡性淋巴瘤、弥散性大b细胞淋巴瘤、套细胞淋巴瘤、肝细胞癌、甲状腺癌、胃癌、头颈癌、小细胞癌、原发性血小板增多症、肌萎缩性骨髓化生、嗜酸性粒细胞增多症、系统性肥大细胞增多症、常见性嗜酸性粒细胞增多症、慢性嗜酸性粒细胞白血病、神经内分泌癌、类癌瘤等。
另外的示例性癌症描述于表6中。
在一个实施方案中,癌症是恶性(或恶化前)血液病。如本文所用,恶性血液病是指造血或淋巴组织的肿瘤,例如影响血液、骨髓或淋巴结的肿瘤。示例性恶性血液病包括但不限于白血病(例如,急性淋巴细胞白血病(all)、急性髓细胞白血病(aml)、慢性淋巴细胞白血病(cll)、慢性髓细胞白血病(cml)、毛细胞白血病、急性单核细胞白血病(amol)、慢性骨髓单核细胞白血病(cmml)、幼年型骨髓单核细胞白血病(jmml)、或大颗粒淋巴细胞白血病)、淋巴瘤(例如,aids-相关淋巴瘤、皮肤t细胞淋巴瘤、霍奇金淋巴瘤(例如,经典霍奇金淋巴瘤或结节性淋巴细胞为主型霍奇金淋巴瘤)、蕈样真菌病、非霍奇金淋巴瘤(例如,b细胞非霍奇金淋巴瘤(例如,伯基特淋巴瘤、小淋巴细胞淋巴瘤(cll/sll)、弥散性大b细胞淋巴瘤、滤泡性淋巴瘤、免疫母细胞性大淋巴瘤、前体b淋巴母细胞淋巴瘤或套细胞淋巴瘤)或t细胞非霍奇金淋巴瘤(蕈样真菌病、间变性大细胞淋巴瘤、或前体t淋巴母细胞淋巴瘤))、原发性中枢神经系统淋巴瘤、塞扎里氏综合征(sézarysyndrome)、瓦尔登斯特伦氏巨球蛋白血症(
在一个实施方案中,靶基因或基因产物或其片段选自表1-4或图3a-4d中描述的任何基因或基因产物。
另外的示例性基因在图3a-4d中示出。
在一个实施方案中,靶基因或基因产物或其片段具有与癌症,例如恶性(或恶化前)血液病相关的取代、插入缺失或拷贝数改变中的一种或多种。示例性基因或基因产物包括但不限于abl1、actb、akt1、akt2、akt3、alk、amer1(fam123b或wtx)、apc、aph1a、ar、araf、arfrp1、arhgap26(graf)arid1a、arid2、asmtl、asxl1、atm、atr、atrx、aurka、aurkb、axin1、axl、b2m、bap1、bard1、bcl10、bcl11b、bcl2、bcl2l2、bcl6、bcl7a、bcor、bcorl1、birc3、blm、braf、brca1、brca2、brd4、brip1(bach1)、brsk1、btg2、btk、btla、c11or、f30(emsy)、cad、card11、cbfb、cbl、ccnd1、ccnd2、ccnd3、ccne1、cct6b、cd22、cd274、(pdl1)、cd36、cd58、cd70、cd79a、cd79b、cdc73、cdh1、cdk12、cdk4、cdk6、cdk8、cdkn1b、cdkn2a、cdkn2b、cdkn2c、cebpa、chd2、chek1、chek2、cic、ciita、cks1b、cps1、crebbp、crkl、crlf2、csf1r、csf3r、ctcf、ctnna1、ctnnb1、cux1、cxcr4、daxx、ddr2、ddx3x、dnm2、dnmt3a、dot1l、dtx1、dusp2、dusp9、ebf1、ect2l、eed、egfr、elp2、ep300、epha3、epha5、epha7、ephb1、erbb2、erbb3、erbb4、erg、esr1、ets1、etv6、exosc6、ezh2、faf1、fam46c、fanca、fancc、fancd2、fance、fancf、fancg、fancl、fas(tnfrsf6)、fbxo11、fbxo31、fbxw7、fgf10、fgf14、fgf19、fgf23、fgf3、fgf4、fgf6、fgfr1、fgfr2、fgfr3、fgfr4、fhit、flcn、flt1、flt3、flt4、flywch1、foxl2、foxo1、foxo3、foxp1、frs2、gadd45b、gata1、gata2、gata3、gid4(c17orf39)、gna11、gna12、gna13、gnaq、gnas、gpr124、grin2a、gsk3b、gtse1、hdac1、hdac4、hdac7、hgf、hist1h1c、hist1h1d、hist1h1e、hist1h2ac、hist1h2ag、hist1h2al、hist1h2am、hist1h2bc、hist1h2bj、hist1h2bk、hist1h2bo、hist1h3b、hnf1a、hras、hsp90aa1、ick、id3、idh1、idh2、igf1r、ikbke、ikzf1、ikzf2、ikzf3、il7r、inhba、inpp4b、inpp5d(ship)、irf1、irf4、irf8、irs2、jak1、jak2、jak3、jarid2、jun、kat6a(myst3)、kdm2b、kdm4c、kdm5a、kdm5c、kdm6a、kdr、keap1、kit、klhl6、kmt2a(mll)、kmt2b(mll2)、kmt2c(mll3)、kras、lef1、lrp1b、lrrk2、maf、mafb、maged1、malt1、map2k1、map2k2、map2k4、map3k1、map3k14、map3k6、map3k7、mapk1、mcl1、mdm2、mdm4、med12、mef2b、mef2c、men1、met、mib1、mitf、mki67、mlh1、mpl、mre11a、msh2、msh3、msh6、mtor、mutyh、myc、mycl(mycl1)、mycn、myd88、myo18a、ncor2、ncstn、nf1、nf2、nfe2l2、nfkbia、nkx2-1、nod1、notch1、notch2、npm1、nras、nt5c2、ntrk1、ntrk2、ntrk3、nup93、nup98、p2ry8、pag1、pak3、palb2、pask、pax5、pbrm1、pc、pcbp1、pclo、pdcd1、pdcd11、pdcd1lg2(pdl2)、pdgfra、pdgfrb、pdk1、phf6、pik3ca、pik3cg、pik3r1、pik3r2、pim1、plcg2、pot1、ppp2r1a、prdm1、prkar1a、prkdc、prss8、ptch1、pten、ptpn11、ptpn2、ptpn6(shp-1)、ptpro、rad21、rad50、rad51、raf1、rara、rasgef1a、rb1、reln、ret、rhoa、rictor、rnf43、ros1、rptor、runx1、s1pr2、sdha、sdhb、sdhc、sdhd、serp2、setbp1、setd2、sf3b1、sgk1、smad2、smad4、smarca1、smarca4、smarcb1、smc1a、smc3、smo、socs1、socs2、socs3、sox10、sox2、spen、spop、src、srsf2、stag2、stat3、stat4、stat5a、stat5b、stat6、stk11、sufu、suz12、taf1、tbl1xr1、tcf3、tcl1a、tet2、tgfbr2、tll2、tmem30a、tmsb4xp8(tmsl3)、tnfaip3、tnfrsf11a、tnfrsf14、tnfrsf17、top1、tp53、tp63、traf2、traf3、traf5、tsc1、tsc2、tshr、tusc3、tyk2、u2af1、u2af2、vhl、wdr90、whsc1(mmset或nsd2)、wisp3、wt1、xbp1、xpo1、yy1ap1、zmym3、znf217、znf24(zscan3)、znf703或zrsr2。
在一个实施方案中,靶基因或基因产物或其片段具有与癌症,例如恶性(或恶化前)血液病相关的一个或多个重排。示例性基因或基因产物包括但不限于alk、bcl6、braf、crlf2、epor、etv4、etv6、fgfr2、igk、bcl2、bcr、ccnd1、egfr、etv1、etv5、ewsr1、igh、igl、jak1、kmt2a(mll)、ntrk1、pdgfrb、rara、ros1、trg、jak2、myc、pdgfra、raf1、ret或tmprss2。
在另一个实施方案中,靶基因或基因产物或其片段具有与癌症相关的一个或多个融合体。示例性基因或基因产物包括但不限于abi1、cbfa2t3、eif4a2、fus、jak1、muc1、pbx1、rnf213、tet1、abl1、cbfb、elf4、gas7、jak2、myb、pcm1、ros1、tfe3、abl2、cbl、ell、gli1、jak3、myc、pcsk7、rpl22、tfg、acsl6、ccnd1、eln、gmps、jazf1、myh11、pdcd1lg2(pdl2)、rpn1、tfpt、aff1、ccnd2、eml4、gphn、kat6a(myst3)、myh9、pde4dip、runx1、tfrc、aff4、ccnd3、ep300、herpud1、kdsr、naca、pdgfb、runx1t1(eto)、tlx1、alk、cd274(pdl1)、epor、hey1、kif5b、nbeap1(bcl8)、pdgfra、runx2、tlx3、arhgap26(graf)、cdk6、eps15、hip1、kmt2a(mll)、ncoa2、pdgfrb、sec31a、tmprss2、arhgef12、cdx2、erbb2、hist1h4i、lasp1、ndrg1、per1、sept5、tnfrsf11a、arid1a、chic2、erg、hlf、lcp1、nf1、phf1、sept6、top1、arnt、chn1、ets1、hmga1、lmo1、nf2、picalm、sept9、tp63、asxl1、cic、etv1、hmga2、lmo2、nfkb2、pim1、set、tpm3、atf1、ciita、etv4、hoxa11、lpp、nin、plag1、sh3gl1、tpm4、atg5、clp1、etv5、hoxa13、lyl1、notch1、pml、slc1a2、trim24、atic、cltc、etv6、hoxa3、maf、npm1、pou2af1、snx29(rundc2a)、trip11、bcl10、cltcl1、ewsr1、hoxa9、mafb、nr4a3、ppp1cb、srsf3、ttl、bcl11a、cntrl(cep110)、fcgr2b、hoxc11、malt1、nsd1、prdm1、ss18、tyk2、bcl11b、col1a1、fcrl4、hoxc13、mds2、ntrk1、prdm16、ssx1、usp6、bcl2、creb3l1、fev、hoxd11、mecom、ntrk2、prrx1、ssx2、whsc1(mmset或nsd2)、bcl3、creb3l2、fgfr1、hoxd13、mkl1、ntrk3、psip1、ssx4、whsc1l1、bcl6、crebbp、fgfr1op、hsp90aa1、mlf1、numa1、ptch1、stat6、ypel5、bcl7a、crlf2、fgfr2、hsp90ab1、mllt1(enl)、nup214、ptk7、stl、zbtb16、bcl9、csf1、fgfr3、igh、mllt10(af10)、nup98、rabep1、syk、zmym2、bcor、ctnnb1、fli1、igk、mllt3、nutm2a、raf1、taf15、znf384、bcr、ddit3、fnbp1、igl、mllt4、(af6)、omd、ralgds、tal1、znf521、birc3、ddx10、foxo1、ikzf1、mllt6、p2ry8、rap1gds1、tal2、braf、ddx6、foxo3、il21r、mn1、pafah1b2、rara、tbl1xr1、btg1、dek、foxo4、il3、mnx1、pax3、rbm15、tcf3(e2a)、camta1、dusp22、foxp1、irf4、msi2、pax5、ret、tcl1a(tcl1)、cars、egfr、fstl3、itk、msn、pax7、rhoh或tec。
另外的示例性基因例如在国际申请公布wo2012/092426的表1-11中描述,所述申请公布的内容以引用的方式整体并入。
上述方法的应用包括使用含有特定基因的所有已知序列变体(或其子集)的寡核苷酸的文库,以用于在医学样本中测序。
在某些实施方案中,方法或测定还包括以下的一种或多种:
(i)对核酸样品进行指纹分析;
(ii)定量核酸样品中的基因或基因产物(例如,本文所述的基因或基因产物)的丰度;
(iii)定量样品中的转录物的相对丰度;
(iv)将核酸样品鉴定为属于特定受试者(例如,正常对照或癌症患者);
(v)鉴定核酸样品中的遗传性状(例如,一个或多个受试者的遗传构成(例如,民族、种族、家族性状));
(vi)确定核酸样品中的倍性;确定核酸样品中的杂合性丢失;
(vii)确定核酸样品中的基因重复事件的存在或不存在;
(viii)确定核酸样品中的基因扩增事件的存在或不存在;或
(ix)确定核酸样品中的肿瘤/正常细胞混合物的水平。
核酸样品
各种各样的组织样品可以是本方法中使用的核酸样品的来源。基因组或亚基因组核酸(例如,dna或rna)可从受试者的样品(例如,肿瘤样品、正常相邻组织(nat)、血液样品)、含有循环肿瘤细胞(ctc)的样品或任何正常对照)中分离出来。在某些实施方案中,将组织样品保存为冷冻的样品或甲醛固定的或多聚甲醛固定的石蜡包埋(ffpe)组织制剂。例如,样品可被包埋在基质中,例如ffpe块或冷冻样品。在某些实施方案中,组织样品是血液样品。在其他实施方案中,组织样品是骨髓抽吸物(bma)样品。分离步骤可包括单个染色体的流式分选;和/或显微解剖受试者的样品(例如,肿瘤样品、nat、血液样品)。
“分离的”核酸分子是与存在于核酸分子的天然来源中的其他核酸分子分开的核酸分子。在某些实施方案中,“分离的”核酸分子在核酸源自其中的生物体的基因组dna中,不含天然位于核酸两侧的序列(诸如蛋白质编码序列)(即,位于核酸的5'端和3'端的序列)。例如,在各种实施方案中,分离的核酸分子在核酸源自其中的细胞的基因组dna中可包含小于约5kb、小于约4kb、小于约3kb、小于约2kb、小于约1kb、小于约0.5kb或小于约0.1kb的核苷酸序列,所述核苷酸序列天然位于核酸分子两侧。此外,“分离的”核酸分子诸如rna分子或cdna分子可基本上不含其他细胞物质或培养基(例如当通过重组技术产生时),或基本上不含化学前体或其他化学品(例如当化学合成时)。
语言“基本上不含其他细胞物质或培养基”包括核酸分子的制剂,其中分子与细胞的细胞组分分开,所述分子从所述细胞中分离或重组产生。因此,基本上不含细胞物质的核酸分子包括具有小于约30%、小于约20%、小于约10%或小于约5%(按干重计)的其他细胞物质或培养基的核酸分子的制剂。
在某些实施方案中,核酸从陈置样品,例如陈置ffpe样品中分离。陈置样品可以例如是有年数的,例如1年、2年、3年、4年、5年、10年、15年、20年、25年、50年、75年、或100年或更久。
核酸样品可从各种大小的组织样品(例如,活组织检查物、ffpe样品、血液样品或骨髓抽吸物样品)中获得。例如,核酸可以从5至200μm或更大的组织样品中分离。例如,组织样品可以测量为5μm、10μm、20μm、30μm、40μm、50μm、70μm、100μm、110μm、120μm、150μm或200μm或更大。
用于从组织样品中进行dna分离的方案是本领域中已知的,例如国际专利申请公布wo2012/092426的实施例1中所提供。另外的从甲醛固定的或多聚甲醛固定的石蜡包埋(ffpe)组织中分离核酸(例如,dna)的方法公开在例如croninm.等,(2004)amjpathol.164(1):35–42;masudan.等,(1999)nucleicacidsres.27(22):4436–4443;spechtk.等,(2001)amjpathol.158(2):419–429,ambionrecoveralltm总核酸分离方案(ambion,登记号am1975,2008年9月)、
用于rna分离的方案公开在例如
分离的核酸样品(例如,基因组dna样品)可通过实施常规技术来进行片段化或剪切。例如,基因组dna可通过物理剪切方法、酶解方法、化学裂解方法和本领域技术人员熟知的其他方法进行片段化。核酸文库可包含所有或基本上所有的基因组复杂性。在这个背景下,术语“基本上所有”是指在程序的初始步骤过程中,实际上可存在一些不希望的基因组复杂性丢失的可能性。本文描述的方法在其中核酸文库是基因组的一部分(即,其中通过设计减少基因组的复杂性)的情况下也是有用的。在一些实施方案中,基因组的任何选择的部分可连同本文描述的方法一起使用。在某些实施方案中,分离整个外显子组或其子集。
本发明特征的方法还可包括分离核酸样品以提供文库(例如,本文所述的核酸文库)。在某些实施方案中,核酸样品包含全基因组片段、亚基因组片段、或两者。分离的核酸样品可用于制备核酸文库。因此,在一个实施方案中,本发明特征的方法还包括分离核酸样品以提供文库(例如,本文所述的核酸文库)。用于从全基因组或亚基因组片段中分离和制备文库的方案是本领域中已知的(例如,illumina的基因组dna样品制备试剂盒)。在某些实施方案中,从受试者的样品(例如,肿瘤样品、正常相邻组织(nat)、血液样品或任何正常对照))中分离基因组或亚基因组dna片段。在一个实施方案中,样品(例如,肿瘤或nat样品)是保存的样本。例如,样品被包埋在基质中,例如ffpe块或冷冻样品。在某些实施方案中,分离步骤包括单个染色体的流式分选;和/或显微解剖受试者的样品(例如,肿瘤样品、nat、血液样品)。在某些实施方案中,用于产生核酸文库的核酸样品小于5微克、小于1微克、或小于500ng、小于200ng、小于100ng、小于50ng、小于10ng、小于5ng、或小于1ng。
在其他实施方案中,用于产生文库的核酸样品包含rna或源自rna的cdna。在一些实施方案中,rna包括总细胞rna。在其他实施方案中,某些丰富的rna序列(例如,核糖体rna)已被耗尽。在一些实施方案中,已富集总rna制剂中的聚(a)尾部mrna级分。在一些实施方案中,cdna通过随机引发的cdna合成方法产生。在其他实施方案中,cdna合成通过由含寡聚(dt)的寡核苷酸引发而开始于成熟mrna的聚(a)尾部。用于耗尽、聚(a)富集和cdna合成的方法是本领域技术人员熟知的。
方法还可包括通过本领域技术人员熟知的特异性或非特异性核酸扩增方法来扩增核酸样品。在一些实施方案中,核酸样品例如通过全基因组扩增方法诸如随机引发的链置换扩增来扩增。
在其他实施方案中,核酸样品通过物理或酶促方法来片段化或剪切,并且连接至合成衔接子,大小选择(例如,通过制备型凝胶电泳)和扩增(例如,通过pcr)。在其他实施方案中,使用片段化和衔接子连接的核酸组而不需要杂交选择前的明确的大小选择或扩增。
在其他实施方案中,分离的dna(例如,基因组dna)是片段化或剪切的。在一些实施方案中,文库包括小于50%的基因组dna,诸如基因组dna的亚组分,所述基因组dna的亚组分是例如已通过其他方式亚组分化(subfractionated)的基因组的减少表示或限定部分。在其他实施方案中,文库包括所有或基本上所有的基因组dna。
在一些实施方案中,文库包括小于50%的基因组dna,诸如基因组dna的亚组分,所述基因组dna的亚组分是例如已通过其他方式亚组分化的基因组的减少表示或限定部分。在其他实施方案中,文库包括所有或基本上所有的基因组dna。用于从全基因组或亚基因组片段中分离和制备文库的方案是本领域中已知的(例如,illumina的基因组dna样品制备试剂盒),并且在本文中描述于实施例中。用于dna剪切的替代方法是本领域中已知的,例如在国际专利申请公布wo2012/092426中的实施例4中所述。例如,替代dna剪切方法可更自动化和/或更有效(例如,对于降解的ffpe样品)。dna剪切方法的替代方案也可用于避免文库制备过程中的连接步骤。
例如当源dna或rna的量是有限时(例如,甚至在全基因组扩增后),可使用少量的核酸执行本文描述的方法。在一个实施方案中,核酸占核酸样品的5μg、4μg、3μg、2μg、1μg、0.8μg、0.7μg、0.6μg、0.5μg或400ng、300ng、200ng、100ng、50ng、10ng、5ng、1ng或更少。例如,人们通常可用50-100ng的基因组dna开始。然而,如果人们在杂交步骤(例如,液相杂交)之前扩增基因组dna,则人们可以更少的基因组dna开始。因此,在杂交(例如,液相杂交)之前扩增基因组dna是可能的,但不是必需的。
用于产生文库的核酸样品还可包含rna或源自rna的cdna。在一些实施方案中,rna包括总细胞rna。在其他实施方案中,某些丰富的rna序列(例如,核糖体rna)已被耗尽。在其他实施方案中,已富集总rna制剂中的聚(a)尾部mrna级分。在一些实施方案中,cdna通过随机引发的cdna合成方法产生。在其他实施方案中,cdna合成通过由含寡聚(dt)的寡核苷酸引发而开始于成熟mrna的聚(a)尾部。用于耗尽、聚(a)富集和cdna合成的方法是本领域技术人员熟知的。
方法还可包括通过本领域技术人员已知的特异性或非特异性核酸扩增方法来扩增核酸样品。核酸样品可例如通过全基因组扩增方法诸如随机引发的链置换扩增来扩增。
核酸样品可通过本文所述的物理或酶促方法来片段化或剪切,并且连接至合成衔接子,大小选择(例如,通过制备型凝胶电泳)和扩增(例如,通过pcr)。使用片段化和衔接子连接的核酸组而不需要杂交选择前的明确的大小选择或扩增。
在一个实施方案中,核酸样品包含来自非癌细胞或非恶性细胞(例如,肿瘤浸润淋巴细胞)的dna、rna(或源自rna的cdna)或两者。在一个实施方案中,核酸样品包含来自非癌细胞或非恶性细胞(例如,肿瘤浸润淋巴细胞)的dna、rna(或源自rna的cdna)或两者,并且不包含或基本上不含来自癌细胞或恶性细胞的dna、rna(或源自rna的cdna)或两者。
在一个实施方案中,核酸样品包含来自癌细胞或恶性细胞的dna、rna(或源自rna的cdna)。在一个实施方案中,核酸样品包含来自癌细胞或恶性细胞的dna、rna(或源自rna的cdna),并且不包含或基本上不含来自非癌细胞或非恶性细胞(例如,肿瘤浸润淋巴细胞)的dna、rna(或源自rna的cdna)或两者。
在一个实施方案中,核酸样品包含来自非癌细胞或非恶性细胞(例如,肿瘤浸润淋巴细胞)的dna、rna(或源自rna的cdna)或两者,以及来自癌细胞或恶性细胞的dna、rna(或源自rna的cdna)或两者。
诱饵的设计和构建
诱饵可以是核酸分子,例如dna或rna分子,其可与靶核酸杂交(例如,互补),从而允许捕获靶核酸。在某些实施方案中,靶核酸是基因组dna分子。在其他实施方案中,靶核酸是rna分子或源自rna分子的cdna分子。在一个实施方案中,诱饵是rna分子。在其他实施方案中,诱饵包含结合实体,例如亲和标签,所述诱饵例如通过与结合实体结合来允许捕获和分离杂交体,所述杂交体由诱饵和与诱饵杂交的核酸形成。在一个实施方案中,诱饵适合于液相杂交。
通常,rna分子被用作诱饵序列。rna-dna双链体比dna-dna双链体更稳定,因此提供对核酸的潜在更好的捕获。
rna诱饵可如本文其他地方所述,使用本领域中已知的方法制备,所述方法包括但不限于使用dna依赖性rna聚合酶的dna分子的从头化学合成和转录。在一个实施方案中,诱饵序列使用已知的核酸扩增方法(诸如pcr),例如使用人dna或汇集的人dna样品作为模板来产生。然后可将寡核苷酸转化为rna诱饵。在一个实施方案中,例如基于将rna聚合酶启动子序列加入至寡核苷酸的一端来使用体外转录。在一个实施方案中,通过例如使用pcr或其他核酸扩增方法来扩增或再扩增诱饵序列,例如通过使每个靶标特异性引物对中的一个引物的尾部具有rna启动子序列,将rna聚合酶启动子序列加入在诱饵的端部处。在一个实施方案中,rna聚合酶是t7聚合酶、sp6聚合酶或t3聚合酶。在一个实施方案中,rna诱饵用标签(例如,亲和标签)标记。在一个实施方案中,rna诱饵通过体外转录,例如使用生物素化的utp来制备。在另一个实施方案中,在不需要生物素的情况下产生rna诱饵,然后使用本领域熟知的方法,诸如补骨脂素交联,将生物素与rna分子交联。在一个实施方案中,rna诱饵是抗rna酶的rna分子,其可例如通过在转录过程中使用修饰的核苷酸以产生抗rna酶降解的rna分子来制备。在一个实施方案中,rna诱饵仅对应于双链dna靶标的一条链。通常,此类rna诱饵不是自补的,并且作为杂交驱动因子更有效。
可从参考序列设计诱饵集合,使得诱饵对于选择参考序列的靶标是最佳的。在一些实施方案中,使用混合碱基(例如,简并)设计诱饵序列。例如,混合碱基可包含在诱饵序列中的常见snp或突变的位置,以便优化诱饵序列以捕获两个等位基因(例如,snp和非snp;突变体和非突变体)。在一些实施方案中,所有已知的序列变异(或其子集)可用多个寡核苷酸诱饵靶向,而不是通过使用混合的简并寡核苷酸。
在某些实施方案中,诱饵集合包括长度为约100个核苷酸与300个核苷酸之间的寡核苷酸(或多个寡核苷酸)。通常,诱饵集合包括长度为约130个核苷酸与230个核苷酸、或约150与200个核苷酸之间的寡核苷酸(或多个寡核苷酸)。在其他实施方案中,诱饵集合包括长度为约300个核苷酸与1000个核苷酸之间的寡核苷酸(或多个寡核苷酸)。
在一些实施方案中,寡核苷酸中的靶成员特异性序列长度为约40与1000个核苷酸、约70与300个核苷酸、约100与200个核苷酸之间,通常长度为约120与170个核苷酸之间。
在一些实施方案中,诱饵集合包括结合实体。结合实体可以是每个诱饵序列上的亲和标签。在一些实施方案中,亲和标签是生物素分子或半抗原。在某些实施方案中,结合实体通过结合配偶体(诸如亲和素分子)或结合半抗原或其抗原结合片段的抗体来允许从杂交混合物中分离诱饵/成员杂交体。
在其他实施方案中,诱饵集合中的寡核苷酸包含相同靶成员序列的正向和反向补体序列,由此具有反向互补成员特异性序列的寡核苷酸也携带反向补体通用尾部。这可产生相同链(即彼此不互补)的rna转录物。
在其他实施方案中,诱饵集合包括在一个或多个位置处包含简并或混合碱基的寡核苷酸。在其他实施方案中,诱饵集合包括存在于单一物种或生物体群落的群体中的多种或基本上所有已知的序列变体。在一个实施方案中,诱饵集合包括存在于人群中的多种或基本上所有已知的序列变体。
在其他实施方案中,诱饵集合包括cdna序列或源自cdna序列。在其他实施方案中,诱饵集合包括扩增产物(例如,pcr产物),所述扩增产物从基因组dna、cdna或克隆dna中扩增。
在其他实施方案中,诱饵集合包括rna分子。在一些实施方案中,所述集合包括化学、酶促修饰或体外转录的rna分子,所述rna分子包括但不限于对rna酶更具有稳定性和抗性的那些。
在其他实施方案中,诱饵通过us2010/0029498和gnirke,a.等(2009)natbiotechnol.27(2):182-189中描述的方法产生,所述专利和参考文献以引用的方式并入本文。例如,生物素化的rna诱饵可通过获得最初在微阵列上合成的合成长寡核苷酸库,并且扩增寡核苷酸以产生诱饵序列来产生。在一些实施方案中,诱饵通过将rna聚合酶启动子序列加入在诱饵序列的一端处,并且使用rna聚合酶合成rna序列来产生。在一个实施方案中,合成寡脱氧核苷酸的文库可从商业供应商(诸如安捷伦科技公司(agilenttechnologies,inc.))获得,并且使用已知的核酸扩增方法来扩增。
因此,提供了一种制备上述诱饵集合的方法。方法包括选择一个或多个靶标特异性诱饵寡核苷酸序列(例如,本文所述的一个或多个突变捕获、参考或对照寡核苷酸序列);获得靶标特异性诱饵寡核苷酸序列库(例如,合成靶标特异性诱饵寡核苷酸序列库,例如通过微阵列合成);并且任选地,扩增寡核苷酸以产生诱饵集合。
在其他实施方案中,方法还包括使用一种或多种生物素化的引物来扩增(例如,通过pcr)寡核苷酸。在一些实施方案中,寡核苷酸在连接至微阵列的每个寡核苷酸端部处包含通用序列。方法还可包括从寡核苷酸中除去通用序列。此类方法还可包括除去寡核苷酸的互补链,使寡核苷酸退火,并且延伸寡核苷酸。在这些实施方案中的一些中,用于扩增(例如,通过pcr)寡核苷酸的方法使用一种或多种生物素化的引物。在一些实施方案中,方法还包括选择扩增的寡核苷酸的大小。
在一个实施方案中,制备rna诱饵集合。方法包括根据本文描述的方法产生诱饵序列集合,将rna聚合酶启动子序列加入在诱饵序列的一端处,并且使用rna聚合酶来合成rna序列。rna聚合酶可选自t7rna聚合酶、sp6rna聚合酶或t3rna聚合酶。在其他实施方案中,通过扩增(例如,通过pcr)诱饵序列,将rna聚合酶启动子序列加入在诱饵序列的端部处。在其中用基因组dna或cdna中的特异性引物对,通过pcr扩增诱饵序列的实施方案中,将rna启动子序列加入至每对中两个特异性引物中的一个的5'端会产生pcr产物,所述pcr产物可使用标准方法转录成rna诱饵。
在其他实施方案中,诱饵集合可使用人dna或汇集的人dna样品作为模板来产生。在此类实施方案中,通过聚合酶链式响应(pcr)来扩增寡核苷酸。在其他实施方案中,通过滚环扩增或超支化滚环扩增来再扩增已被扩增的寡核苷酸。相同的方法也可用于使用人dna或汇集的人dna样品作为模板来产生诱饵序列。相同的方法也可用于使用由其他方法获得的基因组的亚组分来产生诱饵序列,所述其他方法包括但不限于限制性酶切、脉冲场凝胶电泳、流式分选、cscl密度梯度离心、选择性复性动力学、显微解剖染色体制剂以及本领域技术人员已知的其他分级分离方法。
在某些实施方案中,诱饵集合中的诱饵数量小于1,000。在其他实施方案中,诱饵集合中的诱饵数量大于1,000、大于5,000、大于10,000、大于20,000、大于50,000、大于100,000或大于500,000。
诱饵序列的长度可在约70个核苷酸与1000个核苷酸之间。在一个实施方案中,诱饵长度为约100与300个核苷酸、110与200个核苷酸、或120与170个核苷酸之间的长度。除了以上提到的那些之外,可在本文描述的方法中使用长度为约70、80、90、100、110、120、130、140、150、160、170、180、190、200、210、220、230、240、250、300、400、500、600、700、800和900个核苷酸的中等寡核苷酸长度。在一些实施方案中,可使用约70、80、90、100、110、120、130、140、150、160、170、180、190、200、210、220或230个碱基的寡核苷酸。
每个诱饵序列可包含靶标特异性(例如,成员特异性)诱饵序列和一端或两端上的通用尾部。如本文所用,术语“诱饵序列”可以指靶标特异性诱饵序列或包含靶标特异性“诱饵序列”和寡核苷酸的其他核苷酸的整个寡核苷酸。诱饵中的靶标特异性序列的长度为约40个核苷酸与1000个核苷酸之间。在一个实施方案中,靶标特异性序列的长度为约70个核苷酸与300个核苷酸之间。在另一个实施方案中,靶标特异性序列的长度为约100个核苷酸与200个核苷酸之间。在另一个实施方案中,靶标特异性序列的长度为约120个核苷酸与170个核苷酸之间,通常长度为120个核苷酸。除了以上提到的那些之外,也可在本文描述的方法中使用中等长度,诸如长度为约40、50、60、70、80、90、100、110、120、130、140、150、160、170、180、190、200、210、220、230、240、250、300、400、500、600、700、800和900个核苷酸的靶标特异性序列,以及长度为以上提到的长度之间的靶标特异性序列。
在一个实施方案中,诱饵是长度为约50至200个核苷酸(例如,约50、60、80、90、100、110、120、130、140、150、160、170、190或200个核苷酸)的寡聚物(例如,包括rna寡聚物、dna寡聚物、或其组合)。在一个实施方案中,每种诱饵寡聚物包含约120至170个或通常约120个核苷酸,所述核苷酸是靶标特异性诱饵序列。诱饵可在一端或两端处包含另外的非靶标特异性核苷酸序列。可使用另外的核苷酸序列例如以用于pcr扩增或作为诱饵标识符。在某些实施方案中,诱饵另外包含如本文所述的结合实体(例如,捕获标签诸如生物素分子)。结合实体(例如,生物素分子)可例如在诱饵的5’-端、3’-端处或向内(例如,通过并入生物素化的核苷酸)连接至诱饵。在一个实施方案中,生物素分子连接在诱饵的5’-端处。
在一个示例性实施方案中,诱饵是长度为约150个核苷酸的寡核苷酸,所述约150个核苷酸中的120个核苷酸是靶标特异性“诱饵序列”。其他30个核苷酸(例如,每端上15个核苷酸)是用于pcr扩增的通用任意尾部。尾部可以是由用户选择的任何序列。例如,合成寡核苷酸库可包括序列为5’-atcgcaccagcgtgtn120cactgcggctcctca-3’(seqidno:1)的寡核苷酸,其中n120表示靶标特异性诱饵序列。
本文描述的诱饵序列可用于选择外显子和短靶序列。在一个实施方案中,诱饵的长度为约100个核苷酸与300个核苷酸之间。在另一个实施方案中,诱饵的长度为约130个核苷酸与230个核苷酸之间。在另一个实施方案中,诱饵的长度为约150个核苷酸与200个核苷酸之间。例如用于选择外显子和短靶序列的诱饵中的靶标特异性序列的长度为约40个核苷酸与1000个核苷酸之间。在一个实施方案中,靶标特异性序列的长度为约70个核苷酸与300个核苷酸之间。在另一个实施方案中,靶标特异性序列的长度为约100个核苷酸与200个核苷酸之间。在另一个实施方案中,靶标特异性序列的长度为约120个核苷酸与170个核苷酸之间。
在一些实施方案中,长寡核苷酸可最小化捕获靶序列所必需的寡核苷酸的数量。例如,每个外显子可使用一个寡核苷酸。本领域中已知人基因组中蛋白质编码外显子的平均长度和中值长度分别为约164个碱基对和120个碱基对。与较短的诱饵相比,较长的诱饵可更具特异性并且捕获效果更好。因此,每个寡核苷酸诱饵序列的成功率高于短寡核苷酸的成功率。在一个实施方案中,最小诱饵覆盖的序列是一个诱饵的大小(例如,120-170个碱基),例如以用于捕获外显子大小的靶标。在确定诱饵序列的长度时,人们也可考虑到不必要的长诱饵捕获更多与靶标直接相邻的不希望的dna。与较短的寡核苷酸诱饵相比,较长的寡核苷酸诱饵也可对dna样品中靶向区域中的多态性更耐受。通常,诱饵序列源自参考基因组序列。如果实际dna样品中的靶序列偏离参考序列,例如如果它包含单核苷酸多态性(snp),则它可效率较低地与诱饵杂交,因此可能在与诱饵序列杂交的序列中不足或完全不存在。对于较长的合成诱饵分子,由于snp引起的等位基因漏失是不太可能的,因为与20或70个碱基中的单个错配相比,例如120至170个碱基中的单个错配可对杂交体稳定性具有较小的影响,所述20或70个碱基分别是多重扩增和微阵列捕获中的典型诱饵或引物长度。
为了选择与捕获诱饵长度相比较长的靶标,诸如基因组区域,诱饵序列长度通常在与以上提到的短靶标的诱饵相同的大小范围内,除了仅为了最小化相邻序列的靶向的目的,不需要限制最大诱饵序列的大小。可替代地,寡核苷酸可在更宽的窗口(通常600个碱基)上加标题(title)。这种方法可用于捕获比典型的外显子大更多(例如,约500个碱基)的dna片段。因此,选择了更多不希望的侧翼非靶序列。
诱饵合成
诱饵可以是任何类型的寡核苷酸,例如dna或rna。dna或rna诱饵(“寡聚诱饵”)可单独地合成,或可以阵列形式合成,作为dna或rna诱饵集合(“阵列诱饵”)。无论是以阵列形式提供还是作为分离的寡聚物,寡聚诱饵通常是单链的。诱饵可另外包含如本文所述的结合实体(例如,捕获标签诸如生物素分子)。结合实体(例如,生物素分子)可例如在诱饵的5’-端或3’-端处,通常在诱饵的5’-端处连接至诱饵。诱饵集合可通过本领域描述的方法,例如国际专利申请公布wo2012/092426中所述的方法合成。
杂交条件
本发明特征的方法包括使文库(例如,核酸文库)与多个诱饵接触以提供选择的文库捕获的步骤。接触步骤可在液相杂交中实现。在某些实施方案中,方法包括通过另外的一轮或多轮液相杂交来重复杂交步骤。在一些实施方案中,方法还包括使文库捕获受到另外的一轮或多轮与相同或不同诱饵集合进行的液相杂交。本领域中描述了可适用于本文方法的杂交方法,例如国际专利申请公布wo2012/092426中所述。
本发明的另外的实施方案或特征如下:
另一方面,本发明的特征在于一种制备上述诱饵集合的方法。方法包括选择一个或多个靶标特异性诱饵寡核苷酸序列(例如,对应于本文所述的基因或基因产物的受试者间隔(例如,亚基因组间隔、表达的亚基因组间隔、或两者)的任何诱饵序列);获得靶标特异性诱饵寡核苷酸序列库(例如,合成靶标特异性诱饵寡核苷酸序列库,例如通过微阵列合成);并且任选地,扩增寡核苷酸以产生诱饵集合。
另一方面,本发明的特征在于一种用于确定核酸样品中与癌症表型(例如,正或负)相关的改变(例如,本文描述的基因或基因产物中的至少10、20、30、50或更多个改变)的存在或不存在的方法。方法包括根据本文描述的任何方法和诱饵接触基于液相的响应中的样品中的核酸以获得核酸捕获;并且对核酸捕获的全部或子集进行测序(例如,通过下一代测序),从而确定本文描述的基因或基因产物中改变的存在或不存在)。
在某些实施方案中,诱饵集合包括长度为约100个核苷酸与300个核苷酸之间的寡核苷酸(或多个寡核苷酸)。通常,诱饵集合包括长度为约130个核苷酸与230个核苷酸、或约150与200个核苷酸之间的寡核苷酸(或多个寡核苷酸)。在其他实施方案中,诱饵集合包括长度为约300个核苷酸与1000个核苷酸之间的寡核苷酸(或多个寡核苷酸)。
在一些实施方案中,寡核苷酸中的靶成员特异性序列长度为约40与1000个核苷酸、约70与300个核苷酸、约100与200个核苷酸之间,通常长度为约120与170个核苷酸之间。
在一些实施方案中,诱饵集合包括结合实体。结合实体可以是每个诱饵序列上的亲和标签。在一些实施方案中,亲和标签是生物素分子或半抗原。在某些实施方案中,结合实体通过结合配偶体(诸如亲和素分子)或结合半抗原或其抗原结合片段的抗体来允许从杂交混合物中分离诱饵/成员杂交体。
在其他实施方案中,诱饵集合中的寡核苷酸包含相同靶成员序列的正向和反向补体序列,由此具有反向互补成员特异性序列的寡核苷酸也携带反向补体通用尾部。这可产生相同链(即彼此不互补)的rna转录物。
在其他实施方案中,诱饵集合包括在一个或多个位置处包含简并或混合碱基的寡核苷酸。在其他实施方案中,诱饵集合包括存在于单一物种或生物体群落的群体中的多种或基本上所有已知的序列变体。在一个实施方案中,诱饵集合包括存在于人群中的多种或基本上所有已知的序列变体。
在其他实施方案中,诱饵集合包括cdna序列或源自cdna序列。在一个实施方案中,cdna由rna序列制备,例如肿瘤细胞或癌细胞衍生的rna,例如从肿瘤-ffpe样品、血液样品或骨髓抽吸物样品中获得的rna。在其他实施方案中,诱饵集合包括扩增产物(例如,pcr产物),所述扩增产物从基因组dna、cdna或克隆dna中扩增。
在其他实施方案中,诱饵集合包括rna分子。在一些实施方案中,所述集合包括化学、酶促修饰或体外转录的rna分子,所述rna分子包括但不限于对rna酶更具有稳定性和抗性的那些。
在其他实施方案中,诱饵通过us2010/0029498和gnirke,a.等(2009)natbiotechnol.27(2):182-189中描述的方法产生,所述专利和参考文献以引用的方式并入本文。例如,生物素化的rna诱饵可通过获得最初在微阵列上合成的合成长寡核苷酸库,并且扩增寡核苷酸以产生诱饵序列来产生。在一些实施方案中,诱饵通过将rna聚合酶启动子序列加入在诱饵序列的一端处,并且使用rna聚合酶合成rna序列来产生。在一个实施方案中,合成寡脱氧核苷酸的文库可从商业供应商(诸如安捷伦科技公司)获得,并且使用已知的核酸扩增方法来扩增。
因此,提供了一种制备上述诱饵集合的方法。方法包括选择一个或多个靶标特异性诱饵寡核苷酸序列(例如,本文所述的一个或多个突变捕获、参考或对照寡核苷酸序列);获得靶标特异性诱饵寡核苷酸序列库(例如,合成靶标特异性诱饵寡核苷酸序列库,例如通过微阵列合成);并且任选地,扩增寡核苷酸以产生诱饵集合。
在其他实施方案中,方法还包括使用一种或多种生物素化的引物来扩增(例如,通过pcr)寡核苷酸。在一些实施方案中,寡核苷酸在连接至微阵列的每个寡核苷酸端部处包含通用序列。方法还可包括从寡核苷酸中除去通用序列。此类方法还可包括除去寡核苷酸的互补链,使寡核苷酸退火,并且延伸寡核苷酸。在这些实施方案中的一些中,用于扩增(例如,通过pcr)寡核苷酸的方法使用一种或多种生物素化的引物。在一些实施方案中,方法还包括选择扩增的寡核苷酸的大小。
在一个实施方案中,制备rna诱饵集合。方法包括根据本文描述的方法产生诱饵序列集合,将rna聚合酶启动子序列加入在诱饵序列的一端处,并且使用rna聚合酶来合成rna序列。rna聚合酶可选自t7rna聚合酶、sp6rna聚合酶或t3rna聚合酶。在其他实施方案中,通过扩增(例如,通过pcr)诱饵序列,将rna聚合酶启动子序列加入在诱饵序列的端部处。在其中用基因组dna或cdna中的特异性引物对,通过pcr扩增诱饵序列的实施方案中,将rna启动子序列加入至每对中两个特异性引物中的一个的5'端会产生pcr产物,所述pcr产物可使用标准方法转录成rna诱饵。
在其他实施方案中,诱饵集合可使用人dna或汇集的人dna样品作为模板来产生。在此类实施方案中,通过聚合酶链式响应(pcr)来扩增寡核苷酸。在其他实施方案中,通过滚环扩增或超支化滚环扩增来再扩增已被扩增的寡核苷酸。相同的方法也可用于使用人dna或汇集的人dna样品作为模板来产生诱饵序列。相同的方法也可用于使用由其他方法获得的基因组的亚组分来产生诱饵序列,所述其他方法包括但不限于限制性酶切、脉冲场凝胶电泳、流式分选、cscl密度梯度离心、选择性复性动力学、显微解剖染色体制剂以及本领域技术人员已知的其他分级分离方法。
在某些实施方案中,诱饵集合中的诱饵数量小于1,000,例如2、3、4、5、10、50、100、500个诱饵。在其他实施方案中,诱饵集合中的诱饵数量大于1,000、大于5,000、大于10,000、大于20,000、大于50,000、大于100,000或大于500,000。
在某些实施方案中,文库(例如,核酸文库)包括成员集合。如本文所述,文库成员可包括靶成员(例如,肿瘤成员、参考成员和/或对照成员;在本文中又分别称为第一、第二和/或第三成员)。文库的成员可来自单个个体。在实施方案中,文库可包括来自多于一个受试者(例如,2、3、4、5、6、7、8、9、10、20、30或更多个受试者)的成员,例如来自不同受试者的两个或更多个文库可组合以形成具有来自多于一个受试者的成员的文库。在一个实施方案中,受试者是患有癌症或肿瘤或处于患有癌症或肿瘤风险的人。
如本文所用,“成员”或“文库成员”或其他类似术语是指作为文库成员的核酸分子,例如dna或rna。通常,成员是dna分子,例如基因组dna或cdna。成员可以是剪切的基因组dna。在其他实施方案中,成员可以是cdna。在其他实施方案中,成员可以是rna。成员包含来自受试者的序列,并且还可包含并非源自受试者的序列,例如引物或允许标识的序列,例如“条形码”序列。
在另一个实施方案中,本发明特征的方法还包括分离核酸样品以提供文库(例如,本文所述的核酸文库)。在某些实施方案中,核酸样品包含全基因组片段、亚基因组片段、或两者。用于从全基因组或亚基因组片段中分离和制备文库的方案是本领域中已知的(例如,illumina的基因组dna样品制备试剂盒)。在某些实施方案中,从受试者的样品(例如,肿瘤样品、正常相邻组织(nat)、血液样品或任何正常对照))中分离基因组或亚基因组dna片段。在一个实施方案中,样品(例如,肿瘤或nat样品)是保存的。例如,样品被包埋在基质中,例如ffpe块或冷冻样品。在某些实施方案中,分离步骤包括单个染色体的流式分选;和/或显微解剖受试者的样品(例如,肿瘤样品、nat、血液样品)。在某些实施方案中,用于产生核酸文库的核酸样品小于5微克、小于1微克或小于500ng(例如,200ng或更少)。
在其他实施方案中,用于产生文库的核酸样品包含rna或源自rna的cdna。在一些实施方案中,rna包括总细胞rna。在其他实施方案中,某些丰富的rna序列(例如,核糖体rna)已被耗尽。在一些实施方案中,已富集总rna制剂中的聚(a)尾部mrna级分。在一些实施方案中,cdna通过随机引发的cdna合成方法产生。在其他实施方案中,cdna合成通过由含寡聚(dt)的寡核苷酸引发而开始于成熟mrna的聚(a)尾部。用于耗尽、聚(a)富集和cdna合成的方法是本领域技术人员熟知的。
方法还可包括通过本领域技术人员熟知的特异性或非特异性核酸扩增方法来扩增核酸样品。
在一些实施方案中,核酸样品例如通过全基因组扩增方法诸如随机引发的链置换扩增来扩增。
在其他实施方案中,核酸样品通过物理或酶促方法来片段化或剪切,并且连接至合成衔接子,大小选择(例如,通过制备型凝胶电泳)和扩增(例如,通过pcr)。在其他实施方案中,使用片段化和衔接子连接的核酸组而不需要杂交选择前的明确的大小选择或扩增。
在其他实施方案中,分离的dna(例如,基因组dna)是片段化或剪切的。在一些实施方案中,文库包括小于50%的基因组dna,诸如基因组dna的亚组分,所述基因组dna的亚组分是例如已通过其他方式亚组分化的基因组的减少表示或限定部分。在其他实施方案中,文库包括所有或基本上所有的基因组dna。
在某些实施方案中,文库的成员包含亚基因组间隔,所述亚基因组间隔包含基因内区或基因间区。在另一个实施方案中,亚基因组间隔包含外显子或内含子或其片段,通常外显子序列或其片段。在一个实施方案中,受试者间隔包含编码区或非编码区,例如启动子、增强子、5’非翻译区(5’utr)、或3’非翻译区(3’utr)、或其片段。在其他实施方案中,亚基因组间隔包含cdna或其片段(例如,从肿瘤rna(例如,从肿瘤样品例如ffpe-肿瘤样品中提取的rna)中获得的cdna)。在其他实施方案中,亚基因组间隔包含例如本文所述的snp。在其他实施方案中,靶成员包含基因组中的基本上所有的外显子。在其他实施方案中,靶成员包含本文所述的亚基因组间隔,例如来自选择的所关注的基因或基因产物(例如,与本文所述的癌症表型相关的基因或基因产物)的亚基因组间隔,例如外显子。
在一个实施方案中,亚基因组间隔包含体细胞突变、种系突变或两者。在一个实施方案中,亚基因组间隔包含改变,例如点突变或单突变、缺失突变(例如,框内缺失、基因内缺失、全基因缺失)、插入突变(例如,基因内插入)、倒位突变(例如,染色体内倒位)、连接突变、连接的插入突变、倒位的重复突变、串联重复(例如,染色体内串联重复)、易位(例如,染色体易位、非相互易位)、重排、基因拷贝数变化、或其组合。在某些实施方案中,亚基因组间隔构成样品中肿瘤细胞基因组编码区的小于5%、1%、0.5%、0.1%、0.01%、0.001%。在其他实施方案中,亚基因组间隔不涉及疾病,例如与本文所述的癌症表型无关。
本发明特征的方法包括使一个或多个文库(例如,一个或多个核酸文库)与多个诱饵接触以提供选择的核酸子集(例如,文库捕获)的步骤。在一个实施方案中,接触步骤在固相载体例如阵列中实现。用于杂交的合适的固相载体描述在例如albert,t.j.等.(2007)nat.methods4(11):903-5;hodges,e.等.(2007)nat.genet.39(12):1522-7;以及okou,d.t.等.(2007)nat.methods4(11):907-9中,所述参考文献的内容以引用的方式并入本文。在其他实施方案中,接触步骤在液相杂交中实现。在某些实施方案中,方法包括通过另外的一轮或多轮杂交来重复杂交步骤。在一些实施方案中,方法还包括使文库捕获受到另外的一轮或多轮与相同或不同诱饵集合进行的杂交。
在其他实施方案中,本发明特征的方法还包括扩增文库捕获(例如,通过pcr)。在其他实施方案中,不扩增文库捕获。
在其他实施方案中,方法还包括分析文库捕获。在一个实施方案中,通过测序方法,例如本文所述的下一代测序方法来分析文库捕获。方法包括通过液相杂交来分离文库捕获,并且通过核酸测序对文库捕获进行处理。在某些实施方案中,可重新测序文库捕获。下一代测序方法是本领域中已知的,并且描述于例如metzker,m.(2010)naturebiotechnologyreviews11:31-46中。
在其他实施方案中,方法还包括使文库捕获受到基因分型,从而鉴定选择的核酸的基因型的步骤。
在某些实施方案中,方法还包括以下的一种或多种:
i)对核酸样品进行指纹分析;
ii)定量核酸样品中的基因或基因产物(例如,本文所述的基因或基因产物)的丰度(例如,定量样品中的转录物的相对丰度);
iii)将核酸样品鉴定为属于特定受试者(例如,正常对照或癌症患者);
iv)鉴定核酸样品中的遗传性状(例如,一个或多个受试者的遗传构成(例如,民族、种族、家族性状));
v)确定核酸样品中的倍性;确定核酸样品中的杂合性丢失;
vi)确定核酸样品中的基因重复事件的存在或不存在;
vii)确定核酸样品中的基因扩增事件的存在或不存在;或
viii)确定核酸样品中的肿瘤/正常细胞混合物的水平。
本文描述的任何方法可与以下实施方案中的一种或多种组合。
在一个实施方案中,方法包括获取从肿瘤和/或对照核酸样品(例如,ffpe衍生的核酸样品、或源自血液样品或骨髓抽吸物样品的核酸样品)中获得的核苷酸序列读段。
在一个实施方案中,通过下一代测序方法来提供读段。
在一个实施方案中,方法包括提供核酸成员的文库,并且对来自所述文库的多个成员的预选亚基因组间隔进行测序。在实施方案中,方法可包括选择所述文库的子集以用于测序的步骤,例如基于液相的选择。
在某些实施方案中,方法包括杂交捕获方法,所述杂交捕获方法被设计为捕获两种或更多种不同的靶标分类,每种分类都具有不同的诱饵设计策略。杂交捕获方法和组合物意图捕获靶序列(例如,靶成员)的确定子集并且提供靶序列的均匀覆盖,同时最小化所述子集之外的覆盖。在一个实施方案中,靶序列包含基因组dna中的整个外显子组、或其选择的子集。本文公开的方法和组合物提供不同的诱饵集合,以用于实现复杂靶核酸序列(例如,核酸文库)的不同覆盖深度和模式。
在某些实施方案中,不同分类的诱饵集合和靶标如下。
a.第一诱饵集合,其选择高水平靶标(例如,一个或多个肿瘤成员和/或参考成员,诸如基因、外显子或碱基),对于所述高水平靶标,需要最深覆盖以实现对于以低频率出现的突变的高水平灵敏度。例如,检测以约5%或更少的频率出现的点突变(即,5%的细胞在其基因组中具有这种突变,样品从所述细胞中制备)。第一诱饵集合通常需要约500x或更高的测序深度以确保高检测可靠性。在一个实施方案中,第一诱饵集合选择在某些类型的癌症中频繁突变的一个或多个亚基因组间隔(例如,外显子),例如根据表1-4或图3a-4d的基因或基因产物。
b.第二诱饵集合,其选择中等水平靶标(例如,一个或多个肿瘤成员和/或参考成员,诸如基因、外显子或碱基),对于所述中等水平靶标,需要高覆盖度以实现对于以比高水平靶标更高的频率(例如,约10%的频率)出现的突变的高水平灵敏度。例如,检测以10%的频率出现的改变(例如,点突变)需要约200x或更高的测序深度以确保高检测可靠性。在一个实施方案中,第二诱饵集合选择一个或多个亚基因组间隔(例如,外显子),所述一个或多个亚基因组间隔选自根据表1-4或图3a-4d的基因或基因产物。
c.第三诱饵集合,其选择低水平靶标(例如,一个或多个pgx成员,诸如基因、外显子或碱基),对于所述低水平靶标需要低至中等覆盖度以实现例如对检测杂合等位基因的高水平灵敏度。例如,杂合等位基因的检测需要10-100x的测序深度以确保高检测可靠性。在一个实施方案中,第三诱饵集合选择一个或多个亚基因组间隔(例如,外显子),所述一个或多个亚基因组间隔选自:a)可解释患者代谢不同药物的能力的药物基因组学snp,b)可用于对患者进行独特地鉴定(指纹分析)的基因组snp,和c)可用于评价基因组dna的拷贝数增加/减少以及杂合性丢失(loh)的基因组snp/基因座。
d.第四诱饵集合,其选择内含子靶标(例如,内含子成员),对于所述内含子靶标需要低至中等覆盖度以检测结构断点,诸如基因组易位或插入缺失。例如,检测内含子断点需要5-50x的序列对跨越深度,以确保高检测可靠性。所述第四诱饵集合可用于检测例如易于易位/插入缺失的癌症基因。
e.第五诱饵集合,其选择内含子靶标(例如,内含子成员),对于所述内含子靶标需要稀疏的覆盖以提高检测拷贝数变化的能力。例如,检测若干个末端外显子的一拷贝缺失需要0.1-10x覆盖度以确保高检测可靠性。所述第五诱饵集合可用于检测例如易于扩增/缺失的癌症基因。
本发明特征的方法和组合物涉及调整每个诱饵集合/靶标分类的相对序列覆盖度。用于在诱饵设计中实现相对序列覆盖度差异的方法包括以下的一种或多种:
(i)不同诱饵集合的差异表示–捕获给定靶标(例如,靶成员)的诱饵集合设计可包括在更多/更少数量的拷贝中,以增强/减少相对靶标覆盖深度;
(ii)诱饵子集的差异重叠–捕获给定靶标(例如,靶成员)的诱饵集合设计可包括相邻诱饵之间更长或更短的重叠,以增强/减少相对靶标覆盖深度;
(iii)差异诱饵参数–捕获给定靶标(例如,靶成员)的诱饵集合设计可包括序列修饰/长度更短以减小捕获效率并且降低相对靶标覆盖深度;
(iv)不同诱饵集合的混合–设计为捕获不同靶标集合的诱饵集合可在不同摩尔比下混合,以增强/减少相对靶标覆盖深度;
(v)使用不同类型的寡核苷酸诱饵集合–在某些实施方案中,诱饵集合可包括:
(a)一个或多个化学(例如,非酶促)合成的(例如,单独合成的)诱饵,
(b)一个或多个以阵列形式合成的诱饵,
(c)一个或多个酶促制备的,例如体外转录的诱饵;
(d)(a)、(b)和/或(c)的任何组合,
(e)一个或多个dna寡核苷酸(例如,天然或非天然存在的dna寡核苷酸),
(f)一个或多个rna寡核苷酸(例如,天然或非天然存在的rna寡核苷酸),
(g)(e)和(f)的组合,或
(h)上述任何一项的组合。
不同的寡核苷酸组合可在不同比率下混合,例如选自1:1、1:2、1:3、1:4、1:5、1:10、1:20、1:50、1:100、1:1000等的比率。在一个实施方案中,化学合成的诱饵与阵列产生的诱饵的比率选自1:5、1:10或1:20。dna或rna寡核苷酸可以是天然或非天然存在的。在某些实施方案中,诱饵包含一个或多个非天然存在的核苷酸,以例如增加熔解温度。示例性非天然存在的寡核苷酸包括修饰的dna或rna核苷酸。示例性修饰的rna核苷酸是锁核酸(lna),其中lna核苷酸的核糖部分用连接2'氧和4'碳的额外桥修饰(kaur,h;arora,a;wengel,j;maiti,s;arora,a.;wengel,j.;maiti,s.(2006)."thermodynamic,counterion,andhydrationeffectsfortheincorporationoflockednucleicacidnucleotidesintodnaduplexes".biochemistry45(23):7347–55)。其他修饰的示例性dna和rna核苷酸包括但不限于肽核酸(pna),所述肽核酸由通过肽键连接的重复n-(2-氨乙基)-甘氨酸单元组成(egholm,m.等.(1993)nature365(6446):566–8);修饰以捕获低gc区域的dna或rna寡核苷酸;双环核酸(bna)或交联寡核苷酸;修饰的5-甲基脱氧胞苷;以及2,6-二氨基嘌呤。其他修饰的dna和rna核苷酸是本领域中已知的。
在某些实施方案中,获得靶序列(例如,靶成员)的基本上均一或均匀的覆盖。例如,在每个诱饵集合/靶分类中,可通过修改诱饵参数来优化覆盖的均一性,例如,通过以下的一种或多种:
(i)增加/减少诱饵表示或重叠可用于增强/减少靶标(例如,靶成员)的覆盖度,所述靶标相对于相同分类中的其他靶标是欠覆盖/过度覆盖;
(ii)对于低覆盖度,难以捕获靶序列(例如,高gc含量序列),扩大用诱饵集合靶向的区域以覆盖例如相邻序列(例如,富含gc较少的相邻序列);
(iii)修饰诱饵序列可用于减少诱饵的二级结构并且提高其选择效率;
(iv)修改诱饵长度可用于均衡相同分类内不同诱饵的熔解杂交动力学。诱饵长度可直接修改(通过产生具有不同长度的诱饵)或间接修改(通过产生长度一致的诱饵,并且用任意序列替换诱饵端部);
(v)修饰针对相同靶区域具有不同取向的诱饵(即正向和反向链)可具有不同的结合效率。可选择具有为每个靶标提供最佳覆盖度的任一取向的诱饵集合;
(vi)修改存在于每个诱饵上的结合实体例如捕获标签(例如生物素)的量可影响其结合效率。增加/减少靶向特异性靶标的诱饵的标签水平可用于增强/减少相对靶标覆盖度;
(vii)修改用于不同诱饵的核苷酸类型可用于影响对靶标的结合亲和力,并且增强/减少相对靶标覆盖度;或
(viii)使用例如具有更稳定的碱基配对的修饰的寡核苷酸诱饵,可用于均衡低gc含量或正常gc含量的区域相对于高gc含量的区域之间的熔解杂交动力学。
例如,可使用不同类型的寡核苷酸诱饵集合。
在一个实施方案中,通过使用不同类型的诱饵寡核苷酸以涵盖预选的靶区域来修改选择效率的值。例如,第一诱饵集合(例如,包括10,000-50,000个rna或dna诱饵的基于阵列的诱饵集合)可用于覆盖较大的靶区域(例如,总共1-2mb的靶区域)。第一诱饵集合可掺有第二诱饵集合(例如,包括小于5,000个诱饵的单独合成的rna或dna诱饵集合)以覆盖预选的靶区域(例如,选择的靶区域的跨越例如250kb或更少的所关注的亚基因组间隔)和/或具有更高二级结构例如更高gc含量的区域。选择的所关注的受试者间隔可对应于本文描述的基因或基因产物、或其片段中的一种或多种。根据所需的诱饵重叠,第二诱饵集合可包括约2,000-5,000个诱饵。在其他实施方案中,第二诱饵集合可包括掺入到第一诱饵集合中的选择的寡聚诱饵(例如,小于400、200、100、50、40、30、20、10个诱饵)。第二诱饵集合可在单个寡聚诱饵的任何比率下混合。例如,第二诱饵集合可包括呈1:1等摩尔比存在的单个诱饵。可替代地,第二诱饵集合可包括在不同比率(例如,1:5、1:10、1:20)下存在的单个诱饵,例如以优化某些靶标(例如,与其他靶标相比,某些靶标可具有5-10x的第二诱饵)的捕获。
测序
本发明还包括对核酸进行测序的方法。在这些方法中,通过使用本文描述的方法(例如使用液相杂交)来分离核酸文库成员,从而提供文库捕获。可对文库捕获或其亚组进行测序。因此,本发明特征的方法还包括分析文库捕获。在一个实施方案中,通过测序方法,例如本文所述的下一代测序方法来分析文库捕获。方法包括通过液相杂交来分离文库捕获,并且通过核酸测序对文库捕获进行处理。在某些实施方案中,可重新测序文库捕获。
可使用本领域中已知的任何测序方法。对通过选择方法分离的核酸的测序通常使用下一代测序(ngs)来进行。本领域中描述了适用于本文的测序方法,例如国际专利申请公布wo2012/092426中所述。
在已产生ngs读段后,可将它们与已知参考序列比对或从头组装。例如,鉴定样品(例如,肿瘤样品)中的遗传变异诸如单核苷酸多态性和结构变体可通过将ngs读段与参考序列(例如,野生型序列)比对来完成。ngs的序列比对方法描述于例如trapnellc.和salzbergs.l.naturebiotech.,2009,27:455-457中。从头开始组装的实例描述于例如warrenr.等,bioinformatics,2007,23:500-501;butlerj.等,genomeres.,2008,18:810-820;以及zerbinod.r.和birneye.,genomeres.,2008,18:821-829中。可使用来自一个或多个ngs平台的读取数据,例如混合roche/454和illumina/solexa读取数据来执行序列比对或组装。
比对
比对是将读段与位置,例如基因组位置匹配的方法。错位(例如,将来自短读段的碱基对置于基因组中的错误位置上),例如由于实际癌症突变周围的读段的序列上下文(例如,重复序列的存在)引起的错位可导致突变检测的灵敏度降低,因为替代等位基因的读段可移去一大批替代等位基因读段。如果在不存在实际突变的情况下发生有问题的序列上下文,则错位可通过将参考基因组碱基的实际读段置于错误的位置上而引入“突变的”等位基因的人工读段。因为用于多倍多基因分析的突变识别算法应该对甚至低丰度突变具有灵敏度,这些错位可增加假阳性发现率/降低特异性。
如本文所讨论,可通过评估被分析的基因中预期突变位点周围的比对(手动或以自动方式)的质量来解决实际突变的灵敏度降低。待评估的位点可从癌症突变的数据库中获得(例如,cosmic)。被识别为有问题的区域可通过使用被选择以在相关序列上下文中提供更好的性能的算法来补救,例如通过使用较慢但更精确的比对算法(诸如smith-waterman比对)的比对优化(或重新比对)。在一般比对算法无法补救问题的情况下,可创建定制的比对方法,通过例如调整具有含有取代的高可能性的基因的最大差异错配罚分参数;基于某些肿瘤类型中常见的特定突变类型(例如黑色素瘤中的c→t)调整特定的错配罚分参数;或基于某些样品类型中常见的特定突变类型调整特定的错配罚分参数(例如ffpe中常见的取代)。
可通过对测序的样品中的所有突变识别进行手动或自动检查,评价由于错位引起的评估的基因区域中的特异性降低(假阳性率增加)。发现由于错位而易于发生假突变识别的那些区域可受到与上述相同的比对补救措施。在没有发现可能的算法补救措施的情况下,可将来自问题区域的“突变”从测试小组中分类或筛选出去。
本文公开的方法允许使用多种、单独调整的比对方法或算法来优化与重排(例如,插入缺失)相关的亚基因组间隔的测序中的性能,特别是在依赖于对例如来自肿瘤样品的大量不同基因中的大量不同遗传事件进行大规模平行测序的方法中。在实施方案中,使用多种比对方法来分析读段,所述多种比对方法针对不同基因中的许多重排中的每一个单独地定制或调整。在实施方案中,调整可以是以下的(一种或多种的)函数:被测序的基因(或其他亚基因组间隔)、样品中的肿瘤类型、被测序的变体、或样品或受试者的特征。这种选择或使用针对待测序的许多亚基因组间隔精调的比对条件允许优化速度、灵敏度和特异性。当优化相对大量的不同亚基因组间隔的读段比对时,方法特别有效。在实施方案中,方法包括使用针对重排优化的比对方法和针对与重排无关的亚基因组间隔优化的其他比对方法。
因此,在一个实施方案中,本文描述的方法,例如分析肿瘤样品的方法包括本文描述的用于重排的比对方法。
通常,精确检测插入缺失突变是一种比对练习,因为本文中禁用的测序平台上的假插入缺失率相对较低(因此,即使观察到少数正确对准的插入缺失也会是突变的有力证据)。然而,在存在插入缺失的情况下精确比对会是困难的(尤其是当插入缺失长度增加时)。除了与比对相关的一般问题(例如取代)之外,插入缺失本身可导致比对问题。(例如,不能轻易地明确缺失2bp的二核苷酸重复序列。)可通过错误放置较短(<15bp)的明显含有插入缺失的读段来降低灵敏度和特异性。较大的插入缺失(更接近于单个读段的长度,例如36bp的读段)可导致根本无法比对读段,使得在标准的比对读段集合中不可能检测到插入缺失。
癌症突变的数据库可用于解决这些问题并且提高性能。为了减少假阳性插入缺失发现(提高特异性),可检查通常预期的插入缺失周围的区域的由于序列上下文而引起的有问题的比对,并且与上述取代类似地进行处理。为了提高插入缺失检测的灵敏度,可使用若干种不同的方法来使用关于癌症中预期的插入缺失的信息。例如,可模拟包含预期插入缺失的短读段并且尝试比对。可研究比对,并且有问题的插入缺失区域可具有例如通过减小空位开放/延伸罚分或通过比对部分读段(例如读段的前一半或后一半)而调整的比对参数。
可替代地,不仅可用正常参考基因组尝试初始比对,而且可用基因组的替代版本尝试初始比对,所述基因组的替代版本含有每个已知或可能的癌症插入缺失突变。在这种方法中,初始无法比对或错误比对的插入缺失的读段被成功地放置在基因组的替代(突变)版本上。
以这种方式,可针对预期的癌症基因/位点优化插入缺失比对(并且因此识别)。如本文所用,序列比对算法体现了用于从基因组中的哪个位置鉴定读段序列(例如,短读段序列,例如来自下一代测序)的计算方法或方法,所述计算方法或方法最可能源于评价读段序列与参考序列之间的相似性。可将各种算法应用于序列比对问题。一些算法相对慢,但允许相对高的特异性。这些包括例如基于动态编程的算法。动态编程是用于通过将复杂问题分解为更简单的步骤来解决复杂问题的方法。其他方法相对更有效,但通常不那么彻底。这些包括例如设计用于大规模数据库搜索的启发式算法和概率方法。
在比对算法中使用比对参数来调整算法的性能,例如在读段序列与参考序列之间产生最佳的全局或局部比对。比对参数可针对匹配、错配和插入缺失给出权重。例如,较低的权重允许具有更多错配和插入缺失的比对。
序列上下文(例如,重复序列(例如,串联重复、散在重复)的存在)、低复杂区域、插入缺失、假基因或旁系同源物可影响比对特异性(例如,导致错位)。如本文所用,错位是指将来自短读段的碱基对放置在基因组中的错误位置上。
当选择比对算法或基于肿瘤类型(例如,倾向于具有特定突变或突变类型的肿瘤类型)调整比对参数时,可增加比对的灵敏度。
当选择比对算法或基于特定基因类型(例如,癌基因、肿瘤抑制基因)调整比对参数时,可增加比对的灵敏度。不同类型的癌症相关基因中的突变可对癌症表型具有不同的影响。例如,突变癌基因等位基因通常是显性的。突变肿瘤抑制等位基因通常是隐性的,这意味着在大多数情况下,肿瘤抑制基因的两个等位基因必须受到影响,之后表现出效果。
当选择比对算法或基于突变类型(例如,单核苷酸多态性、插入缺失(插入或缺失)、倒位、易位、串联重复)调整比对参数时,可调整(例如,增加)比对的灵敏度。
当选择比对算法或基于突变位点(例如,突变热点)调整比对参数时,可调整(例如,增加)比对的灵敏度。突变热点是指基因组中突变发生频率比正常突变率高100倍的位点。
当选择比对算法或基于样品类型(例如,ffpe样品)调整比对参数时,可调整(例如,增加)比对的灵敏度/特异性。
可基于样品类型(例如,ffpe样品、血液样品或骨髓抽吸物样品),选择比对算法以调整(例如,增加)比对灵敏度/特异性。
在本领域中描述了比对的优化,例如国际专利申请公布wo2012/092426中所述。
突变识别
碱基识别是指测序装置的原始输出。突变识别是指为被测序的核苷酸位置选择核苷酸值例如a、g、t或c的方法。通常,位置的测序读段(或碱基识别)将提供多于一个值,例如一些读段将给出t而一些将给出g。突变识别是将核苷酸值,例如那些值中的一个分配给序列的方法。虽然它被称为“突变”识别,但它可应用于将核苷酸值分配给任何核苷酸位置,例如对应于突变等位基因、野生型等位基因、尚未被表征为突变或野生型的等位基因的位置,或分配给不通过变异性表征的位置。用于突变识别的方法可包括以下的一种或多种:基于参考序列中每个位置处的信息进行独立识别(例如,检查序列读段;检查碱基识别和质量得分;在给定潜在基因型的情况下计算观察到的碱基和质量得分的概率;并且分配基因型(例如,使用贝叶斯规则));除去假阳性(例如,使用深度阈值来排除远低于或高于期望的读取深度的snp;局部重新比对以除去由于小插入缺失引起的假阳性);并且执行基于连锁不平衡(ld)/插补的分析以改进识别。
计算与特定基因型和位置相关的基因型可能性的方程描述于例如lih.和durbinr.bioinformatics,2010;26(5):589-95中。当评估来自所述癌症类型的样品时,可使用针对某种癌症类型中的特定突变的先验期望值。这种可能性可源自癌症突变的公共数据库,例如癌症体细胞突变目录(cosmic)、hgmd(人基因突变数据库)、snp联合、乳腺癌突变数据库(bic)和乳腺癌基因数据库(bcgd)。
基于ld/插补的分析的实例描述于例如browningbl和yuz.am.j.hum.genet.2009,85(6):847-61中。低覆盖snp识别方法的实例描述于例如liy.等,annu.rev.genomicshum.genet.2009,10:387-406中。
比对后,可使用识别方法(例如,贝叶斯突变识别方法)执行对取代的检测;所述识别方法可应用于待评估的基因的每个亚基因组间隔(例如,外显子)中的每个碱基,其中观察到存在替代等位基因。这种方法将在存在突变的情况下观察到读取数据的概率与在单独存在碱基识别错误的情况下观察到读取数据的概率进行比较。如果这种比较足以强烈支持突变的存在,则可识别突变。
已开发了解决针对癌症dna分析的50%或100%频率的极限偏差的方法。(例如,snvmix-bioinformatics.2010年3月15日;26(6):730–736。)然而,本文公开的方法允许考虑在样品dna的1%与100%之间的在任何地方存在突变等位基因的概率,尤其是在低于50%的水平下。这种方法对于检测天然(多克隆)肿瘤dna的低纯度ffpe样品中的突变特别重要。
贝叶斯突变检测方法的一个优点在于:突变存在概率与单独碱基识别错误概率的比较可通过位点处突变存在的先验期望值来加权。如果在针对给定癌症类型的频繁突变位点处观察到替代等位基因的一些读段,则即使突变的证据量不符合通常的阈值,也可确信地识别突变存在。然后,这种灵活性可用于增加甚至更罕见的突变/更低纯度样品的检测灵敏度,或使测试对于读取覆盖度的降低更加稳健。基因组中随机碱基对在癌症中突变的可能性为大约1e-6。在典型的多基因癌症基因组小组中许多位点处发生特定突变的可能性可高出几个数量级。这些可能性可源自癌症突变的公共数据库(例如,cosmic)。插入缺失识别是通过插入或缺失在测序数据中找到与参考序列不同的碱基的方法,通常包括相关的置信度得分或统计学证据度量。
插入缺失识别的方法可包括鉴定候选插入缺失,通过局部重新比对计算基因型可能性以及执行基于ld的基因型推断和识别的步骤。通常,贝叶斯方法用于获得潜在的插入缺失候选物,然后在贝叶斯框架中与参考序列一起测试这些候选物。
产生候选插入缺失的算法描述于例如mckennaa.等,genomeres.2010;20(9):1297-303;yek.等,bioinformatics,2009;25(21):2865-71;lunterg.和goodsonm.genomeres.2010,epubaheadofprint;以及lih.等,bioinformatics2009,bioinformatics25(16):2078-9中。
用于产生插入缺失识别和单个水平基因型可能性的方法包括例如dindel算法(albersc.a.等,genomeres.2011;21(6):961-73)。例如,贝叶斯em算法可用于分析读段,进行初始插入缺失识别,并且针对每个候选插入缺失,产生基因型可能性,然后使用例如qcall(les.q.和durbinr.genomeres.2011;21(6):952-60)进行基因型插补。可基于插入缺失的大小或位置,调整(例如,增加或减少)参数,诸如观察到插入缺失的先验期望值。
在本领域中描述了突变识别的优化,例如国际专利申请公布wo2012/092426中所述。
sgz算法
各种类型的改变例如体细胞改变和种系突变可通过本文描述的方法(例如,测序、比对或突变识别方法)来检测。在某些实施方案中,通过使用sgz算法的方法来进一步鉴定种系突变。sgz算法描述于sun等cancerresearch2014;74(19s):1893-1893;国际申请公布wo2014/183078和美国申请公布2014/0336996中,所述参考文献和申请公布的内容以引用的方式整体并入。
其他实施方案
在本文描述的方法的实施方案中,方法中的步骤或参数用于修改方法中的下游步骤或参数。
在一个实施方案中,肿瘤样品的特征用于修改以下的一种或多种或所有中的下游步骤或参数:从所述样品中分离核酸;文库构建;诱饵设计或选择;杂交条件;测序;读段作图;选择突变识别方法;突变识别;或突变注释。
在一个实施方案中,分离的肿瘤(或对照)核酸的特征用于修改以下的一种或多种或所有中的下游步骤或参数:从所述样品中分离核酸;文库构建;诱饵设计或选择;杂交条件;测序;读段作图;选择突变识别方法;突变识别;或突变注释。
在一个实施方案中,文库的特征用于修改以下的一种或多种或所有中的下游步骤或参数:从所述样品中重新分离核酸;后续的文库构建;诱饵设计或选择;杂交条件;测序;读段作图;选择突变识别方法;突变识别;或突变注释。
在一个实施方案中,文库捕获的特征用于修改以下的一种或多种或所有中的下游步骤或参数:从所述样品中重新分离核酸;后续的文库构建;诱饵设计或选择;杂交条件;测序;读段作图;选择突变识别方法;突变识别;或突变注释。
在一个实施方案中,测序方法的特征用于修改以下的一种或多种或所有中的下游步骤或参数:从所述样品中重新分离核酸;后续的文库构建;诱饵设计或选择;后续的杂交条件确定;后续的测序;读段作图;选择变异识别方法;突变识别;或突变注释。
在一个实施方案中,作图的读段集合的特征用于修改以下的一种或多种或所有中的下游步骤或参数:从所述样品中重新分离核酸;后续的文库构建;诱饵设计或选择;后续的杂交条件确定;后续的测序;后续的读段作图;选择突变识别方法;突变识别;或突变注释。
在一个实施方案中,方法包括获取肿瘤样品特征的值,例如获取以下的值:所述样品中肿瘤细胞的比例;所述肿瘤样品的细胞结构;或来自肿瘤样品的图像。
在实施方案中,方法包括响应于所述获取的肿瘤样品特征的值,选择用于以下的参数:从肿瘤样品中分离核酸,文库构建;诱饵设计或选择;诱饵/文库成员杂交;测序;或突变识别。
在一个实施方案中,方法还包括获取所述肿瘤样品中存在的肿瘤组织的量的值,将所述获取的值与参考标准进行比较,并且如果符合所述参考标准,则接受所述肿瘤样品,例如如果所述肿瘤样品包含大于30%、40%或50%的肿瘤细胞,则接受所述肿瘤样品。
在一个实施方案中,方法还包括例如通过从所述肿瘤样品,从不符合参考标准的肿瘤样品中大体解剖肿瘤组织来获取富含肿瘤细胞的子样品。
在一个实施方案中,方法还包括确定基本对照例如血液样品是否可用,并且如果是,则从所述基本对照中分离对照核酸(例如,dna)。
在一个实施方案中,方法还包括确定nat是否存在于所述肿瘤样品(例如,其中没有基本对照样品可用)中。
在一个实施方案中,方法还包括例如通过从不伴有基本对照的肿瘤样品中的所述nat中大体解剖非肿瘤组织来获取富含非肿瘤细胞的子样品。
在一个实施方案中,方法还包括确定没有基本对照和没有nat可用并且标记所述肿瘤样品以用于分析而不需要匹配的对照。
在一个实施方案中,方法还包括从所述肿瘤样品中分离核酸以提供分离的肿瘤核酸样品。
在一个实施方案中,方法还包括从对照中分离核酸以提供分离的对照核酸样品。
在一个实施方案中,方法还包括丢弃不具有可检测核酸的样品。
在一个实施方案中,方法还包括获取所述分离的核酸样品中核酸产量的值,并且将获取的值与参考标准进行比较,例如,其中如果所述获取的值小于所述参考标准,则在文库构建之前扩增所述分离的核酸样品。
在一个实施方案中,方法还包括获取所述分离的核酸样品中核酸片段大小的值,并且将获取的值与参考标准(例如,至少300、600或900bp的大小,例如平均大小)进行比较。可响应于这种确定来调整或选择本文描述的参数。
在一个实施方案中,方法还包括获得文库,其中文库中所述核酸片段的大小小于或等于参考值,并且在dna分离与制备文库之间不需要片段化步骤的情况下制备所述文库。
在一个实施方案中,方法还包括获取核酸片段,并且如果所述核酸片段的大小等于或大于参考值并且是片段化的,则将此类核酸片段制成文库。
在一个实施方案中,方法还包括例如通过将可鉴定的相异核酸序列(条形码)加入至多个成员中的每一个来标记多个文库成员中的每一个。
在一个实施方案中,方法还包括将引物连接至多个文库成员中的每一个。
在一个实施方案中,方法还包括提供多个诱饵并且
选择多个诱饵,所述选择响应于:1)患者特征,例如年龄、肿瘤时期、既往治疗或耐药性;2)肿瘤类型;3)肿瘤样品的特征;4)对照样品的特征;5)对照的存在或类型;6)分离的肿瘤(或对照)核酸样品的特征;7)文库特征;8)已知与肿瘤样品中的肿瘤类型相关的突变;9)未知与肿瘤样品中的肿瘤类型相关的突变;10)测序(或杂交或覆盖)预选序列或鉴定预选突变的能力,例如与具有高gc区域或重排的序列相关的困难;或11)被测序的基因。
在一个实施方案中,方法还包括例如响应于确定所述肿瘤样品中的低数量的肿瘤细胞,选择诱饵或多个诱饵,与第二基因的成员相比,给予来自第一基因的成员的相对高效的捕获,例如其中第一基因中的突变与肿瘤样品的肿瘤类型的肿瘤表型相关。
在一个实施方案中,方法还包括获取文库捕获特征(例如核酸浓度或表示)的值,并且将获取的值与核酸浓度或表示的参考标准进行比较。
在一个实施方案中,方法还包括选择具有不符合参考标准的文库特征的值的文库以用于返工(例如,用于改变值以符合参考标准)。
在一个实施方案中,方法还包括选择具有符合参考标准的文库特征的值的文库以用于文库定量。
在一个实施方案中,方法还包括为受试者提供肿瘤类型、基因和遗传改变(tga)的关联性。
在一个实施方案中,方法还包括提供具有多个元件的预选数据库,其中每个元件包含tga。
在一个实施方案中,方法还包括表征受试者的tga,所述表征包括:确定所述tga是否存在于预选数据库中,例如经过验证的tga数据库;将来自预选数据库的tga的信息与来自所述受试者的所述tga(注释)相关联;并且任选地,确定所述受试者的第二或后续tga是否存在于所述预选数据库中,并且如果是,则将来自预选数据库的第二或后续tga的信息与存在于所述患者中的所述第二tga相关联。
在一个实施方案中,方法还包括记忆受试者的tga的存在或不存在(并且任选地相关注释)以形成报告。
在一个实施方案中,方法还包括将所述报告传输至接收方。
在一个实施方案中,方法还包括表征受试者的tga,所述表征包括:确定所述tga是否存在于预选数据库中,例如经过验证的tga数据库;或确定不在所述预选数据库中的tga是否具有已知的临床相关g或a,并且如果是,则在所述预选数据库中提供所述tga的条目。
在一个实施方案中,方法还包括记忆在来自受试者的肿瘤样品的dna中发现的突变的存在或不存在以形成报告。
在一个实施方案中,方法还包括记忆受试者的tga的存在或不存在(并且任选地相关注释)以形成报告。
在一个实施方案中,方法还包括将所述报告传输至接收方。
本发明可在任何以下编号的段落中进行定义:
1.一种评估样品(例如,肿瘤样品或源自肿瘤的样品)中的肿瘤突变负荷的方法,方法包括:
a)从样品中提供亚基因组间隔(例如,编码亚基因组间隔)集合的序列,例如核苷酸序列,其中亚基因组间隔集合来自预定的基因集合;以及
b)确定肿瘤突变负荷的值,其中值是亚基因组间隔集合中体细胞改变(例如,一个或多个体细胞改变)的数量的函数,其中所述改变的数量排除:
(i)亚基因组间隔中的功能改变;以及
(ii)亚基因组间隔中的种系改变,
从而评估样品中的肿瘤突变负荷。
2.一种评估样品(例如,肿瘤样品或源自肿瘤的样品)中的肿瘤突变负荷的方法,方法包括:
(i)从样品中获取包括多个肿瘤成员的文库;
(ii)使文库与诱饵集合接触以提供选择的肿瘤成员,其中所述诱饵集合与肿瘤成员杂交,从而提供文库捕获;
(iii)例如通过下一代测序方法,从所述文库捕获中获取来自肿瘤成员的包含改变(例如,体细胞改变)的亚基因组间隔(例如,编码亚基因组间隔)的读段;
(iv)通过比对方法来比对所述读段;
(v)将来自所述读段的核苷酸值分配给预选的核苷酸位置;
(vi)从分配的核苷酸位置集合中选择亚基因组间隔集合,其中亚基因组间隔集合来自预定的基因集合;以及
(vii)确定肿瘤突变负荷的值,其中值是亚基因组间隔集合中体细胞改变(例如,一个或多个体细胞改变)的数量的函数,其中所述改变的数量排除:
(a)亚基因组间隔中的功能改变;以及
(b)亚基因组间隔中的种系改变,
从而评估样品中的肿瘤突变负荷。
3.如权利要求1或2的方法,其中预定的基因集合不包括整个基因组或整个外显子组。
4.如权利要求1-3中任一项的方法,其中亚基因组间隔集合不包括整个基因组或整个外显子组。
5.如权利要求1-4中任一项的方法,其中值表示为预定的基因集合,例如预定的基因集合的编码区的函数。
6.如权利要求1-5中任一项的方法,其中值表示为测序的亚基因组间隔,例如测序的编码亚基因组间隔的函数。
7.如权利要求1-6中任一项的方法,其中值表示为每个预选单位的体细胞改变的数量的函数,例如表示为每兆碱基的体细胞改变的数量的函数。
8.如权利要求1-7中任一项的方法,其中值表示为预定的基因集合,例如预定的基因集合的编码区的预选数量的位置中的体细胞改变的数量的函数。
9.如权利要求1-8中任一项的方法,其中值表示为测序的亚基因组间隔(例如,编码亚基因组间隔)的预选数量的位置中的体细胞改变的数量的函数。
10.如权利要求1-9中任一项的方法,其中值表示为预定的基因集合,例如预定的基因集合的编码区中每兆碱基的体细胞改变的数量的函数。
11.如权利要求1-10中任一项的方法,其中值表示为测序的亚基因组间隔(例如,编码亚基因组间隔)中每兆碱基的改变的数量的函数。
12.如权利要求1-11中任一项的方法,其中将肿瘤突变负荷外推至较大部分的基因组,例如外推至整个外显子组或整个基因组。
13.如权利要求1-12中任一项的方法,其中样品来自受试者,例如患有癌症的受试者,或正在接受或已接受疗法的受试者。
14.如权利要求1-13中任一项的方法,肿瘤突变负荷表示为例如来自参考群体的样品中的肿瘤突变负荷之中的百分位数,所述参考群体例如与受试者患有相同类型癌症的患者、或正在接受或已接受与受试者相同类型疗法的患者的参考群体。
15.如权利要求1-14中任一项的方法,其中功能改变是与参考序列例如野生型或未突变的序列相比,对细胞分裂、生长或存活具有影响例如促进细胞分裂、生长或存活的改变。
16.如权利要求1-15中任一项的方法,其中功能改变本身通过包括在功能改变的数据库例如cosmic数据库(cancer.sanger.ac.uk/cosmic;forbes等nucl.acidsres.2015;43(d1):d805-d811)中来鉴定。
17.如权利要求1-16中任一项的方法,其中功能改变是具有已知功能状态,例如作为cosmic数据库中已知体细胞改变而存在的改变。
18.如权利要求1-17中任一项的方法,其中功能改变是具有可能的功能状态,例如肿瘤抑制基因中的截短的改变。
19.如权利要求1-18中任一项的方法,其中功能改变是司机突变,例如,例如通过增加细胞存活或繁殖而为其微环境中的克隆提供选择性优势的改变。
20.如权利要求1-19中任一项的方法,其中功能改变是能够引起克隆扩增的改变。
21.如权利要求1-20中任一项的方法,其中功能改变是能够引起以下的一种或多种的改变:
(a)生长信号自给自足;
(b)抗生长信号减少,例如对抗生长信号不灵敏;
(c)细胞凋亡减少;
(d)复制潜力增加;
(e)持续的血管生成;或
(f)组织浸润或转移。
22.如权利要求1-21中任一项的方法,其中功能改变不是乘客突变,例如是对克隆的适应性具有可检测影响的改变。
23.如权利要求1-22中任一项的方法,其中功能改变不是意义不明变体(vus),例如不是其致病性既不能被确认也不能被排除的改变。
24.如权利要求1-23中任一项的方法,其中排除预定的基因集合中的预选基因(例如,肿瘤基因)中的多个(例如,10%、20%、30%、40%、50%或75%或更多)功能改变。
25.如权利要求1-24中任一项的方法,其中排除预定的基因集合中的预选基因(例如,肿瘤基因)中的所有功能改变。
26.如权利要求1-25中任一项的方法,其中排除预定的基因集合中的多个预选基因(例如,肿瘤基因)中的多个功能改变。
27.如权利要求1-26中任一项的方法,其中排除预定的基因集合中的所有基因(例如,肿瘤基因)中的所有功能改变。
28.如权利要求1-27中任一项的方法,其中通过使用一种方法来排除种系改变,所述方法不使用与匹配的正常序列比较。
29.如权利要求1-28中任一项的方法,其中通过包括使用sgz算法的方法来排除种系改变。
30.如权利要求1-29中任一项的方法,其中种系改变本身通过包括在种系改变的数据库例如dbsnp数据库(www.ncbi.nlm.nih.gov/snp/index.html;sherry等nucleicacidsres.2001;29(1):308-311)中来鉴定。
31.如权利要求1-30中任一项的方法,其中种系改变本身通过包括在exac数据库的两个或更多个计数(exac.broadinstitute.org;exomeaggregationconsortium等.“analysisofprotein-codinggeneticvariationin60,706humans,”biorxivpreprint.2015年10月30日)中来鉴定。
32.如权利要求1-31中任一项的方法,其中种系改变是单核苷酸多态性(snp)、碱基取代、插入缺失或沉默突变(例如,同义突变)。
33.如权利要求1-32中任一项的方法,其中种系改变本身通过包括在千人基因组计划数据库(www.1000genomes.org;mcvean等nature.2012;491,56–65)中来鉴定。
34.如权利要求1-33中任一项的方法,其中种系改变本身通过包括在esp数据库(外显子组变异数据库,nhlbigo外显子组测序计划(esp),seattle,wa(evs.gs.washington.edu/evs/)中来鉴定。
35.如权利要求1-34中任一项的方法,其中体细胞改变是沉默突变,例如同义改变。
36.如权利要求1-35中任一项的方法,其中体细胞改变是乘客突变,例如对克隆的适应性具有不可检测影响的改变。
37.如权利要求1-36中任一项的方法,其中体细胞改变是意义不明变体(vus),例如其致病性既不能被确认也不能被排除的改变。
38.如权利要求1-37中任一项的方法,其中体细胞改变是点突变。
39.如权利要求1-38中任一项的方法,其中体细胞改变是短变体(例如,短编码变体),例如碱基取代、插入缺失、插入或缺失。
40.如权利要求1-39中任一项的方法,其中体细胞改变是非同义单核苷酸变体(snv)。
41.如权利要求1-40中任一项的方法,其中体细胞改变是剪接变体。
42.如权利要求1-41中任一项的方法,其中体细胞改变尚未被鉴定为与癌症表型相关。
43.如权利要求1-42中任一项的方法,其中体细胞改变不是重排,例如不是易位。
44.如权利要求1-43中任一项的方法,其中预定的基因集合包括多个基因,所述多个基因以突变形式与对细胞分裂、生长或存活的影响相关,或与癌症相关。
45.如权利要求1-44中任一项的方法,其中预定的基因集合包括至少约50或更多、约100或更多、约150或更多、约200或更多、约250或更多、约300或更多、约350或更多、约400或更多、约450或更多、或约500或更多个基因。
46.如权利要求1-45中任一项的方法,其中预定的基因集合包括选自表1-4或图3a-4d的至少约50或更多、约100或更多、约150或更多、约200或更多、约250或更多、约300或更多个、或所有的基因或基因产物。
47.如权利要求1-46中任一项的方法,其还包括从肿瘤样品中获取包括多个肿瘤成员的文库。
48.如权利要求1-47中任一项的方法,其还包括使文库与诱饵集合接触以提供选择的肿瘤成员,其中所述诱饵集合与肿瘤成员杂交,从而提供文库捕获。
49.如权利要求1-48中任一项的方法,其还包括例如通过下一代测序方法,从所述文库或文库捕获中获取来自肿瘤成员的包含体细胞改变的亚基因组间隔的读段,从而获取所述亚基因组间隔的读段。
50.如权利要求1-49中任一项的方法,其还包括通过比对方法来比对所述读段。
51.如权利要求1-50中任一项的方法,其还包括将来自所述读段的核苷酸值分配给预选的核苷酸位置。
52.如权利要求1-51中任一项的方法,其中获取亚基因组间隔的读段包括对亚基因组间隔进行测序,所述亚基因组间隔来自选自表1-4或图3a-4d的至少约50或更多、约100或更多、约150或更多、约200或更多、约250或更多、约300或更多个、或所有的基因或基因产物。
53.如权利要求1-52中任一项的方法,其中获取亚基因组间隔的读段包括用大于约250x、大于约500x、或大于约1,000x的平均独特覆盖度进行测序。
54.如权利要求1-53中任一项的方法,其中获取亚基因组间隔的读段包括在测序的大于95%、大于约97%、或大于约99%的基因(例如,外显子)处用大于约250x、大于约500x、或大于约1,000x的平均独特覆盖度进行测序。
55.如权利要求1-54中任一项的方法,其中序列由权利要求1-54中任一项的方法提供。
56.如权利要求1-55中任一项的方法,其还包括通过以下表征肿瘤样品中的变体,例如改变:
a)获取:
i)序列覆盖度输入(sci),其对于多个选择的亚基因组间隔中的每一个包括在选择的亚基因组间隔处归一化的序列覆盖度的值,其中sci是亚基因组间隔的读段数量和方法匹配的对照的读段数量的函数;
ii)snp等位基因频率输入(safi),其对于多个选择的种系snp中的每一个包括肿瘤样品中等位基因频率的值,其中safi至少部分地基于肿瘤样品中次要或替代等位基因频率;以及
iii)变体等位基因频率输入(vafi),其包括肿瘤样品中所述变体的等位基因频率;
b)获取作为sci和safi的函数的以下值:
i)多个基因组区段中的每一个的基因组区段总拷贝数(c);
ii)多个基因组区段中的每一个的基因组区段次要等位基因拷贝数(m);以及
iii)样品纯度(p),
其中c、m和p的值通过将全基因组拷贝数模型拟合至sci和safi来获得;以及
c)获取:
突变类型g的值,其表示变体是体细胞的、亚克隆体细胞变体、种系的或不可辨识的,并且是vafi、p、c和m的函数。
57.如权利要求1-56中任一项的方法,其还包括对多个选择的亚基因组间隔中的每一个、多个选择的种系snp中的每一个和变体(例如,改变)进行测序,其中归一化之前的平均序列覆盖度为至少约250x,例如至少约500x。
58.如权利要求56或57的方法,其中将全基因组拷贝数模型拟合至sci包括使用以下方程:
59.如权利要求56-58中任一项的方法,其中将全基因组拷贝数模型拟合至safi包括使用以下方程:
60.如权利要求56-59中任一项的方法,其中通过确定vafi、p、c和m的值与体细胞/种系状态的模型的拟合来确定g。
61.如权利要求56-60中任一项的方法,其中通过以下来获取g的值:
62.如权利要求56-61中任一项的方法,其中,
为0或接近0的g值表示变体是体细胞变体;
为1或接近1的g值表示变体是种系变体;
小于1但大于0的g值表示不能辨识的结果;并且
显著小于0的g值表示变体是亚克隆体细胞变体。
63.如权利要求1-62中任一项的方法,其中样品(例如,肿瘤样品或源自肿瘤的样品)包含一个或多个恶化前或恶性细胞;来自实体瘤、软组织肿瘤或转移病变的细胞;来自手术切缘的组织或细胞;组织学正常的组织;一个或多个循环肿瘤细胞(ctc);正常相邻组织(nat);来自患有肿瘤或处于患有肿瘤风险的相同受试者的血液样品;或ffpe样品。
64.如权利要求1-63中任一项的方法,其中样品是ffpe样品。
65.如权利要求63或64的方法,其中ffpe样品具有以下特性中的一种、两种或所有:
(a)表面积为25mm2或更大;
(b)样品体积为1mm3或更大;或
(c)有核细胞结构为80%或更多或30,000个细胞或更多。
66.如权利要求1-65中任一项的方法,其中样品是包含循环肿瘤dna(ctdna)的样品。
67.如权利要求1-66中任一项的方法,其中样品从实体瘤、血液学癌症或其转移形式中获取。
68.如权利要求1-67中任一项的方法,其还包括响应于肿瘤突变负荷的评估,对肿瘤样品或肿瘤样品来自其中的受试者进行分类。
69.如权利要求1-68中任一项的方法,其还包括向患者或向另一个人或主体、护理人员、医生、肿瘤学家、医院、诊所、第三方付款人、保险公司或政府办公室生成报告,例如电子的、基于网络的或纸质的报告。
70.如权利要求69的方法,其中所述报告包括来自方法的输出,所述输出包括肿瘤突变负荷。
71.一种用于评估样品(肿瘤样品或源自肿瘤的样品)中的肿瘤突变负荷的系统,所述系统包括:
可操作地连接至存储器的至少一个处理器,当执行时,至少一个处理器被配置为:
a)从肿瘤样品中获取亚基因组间隔(例如,编码亚基因组间隔)集合的序列,例如核苷酸序列,其中编码亚基因组间隔集合来自预定的基因集合;以及
b)确定肿瘤突变负荷的值,其中值是亚基因组间隔集合中体细胞改变(例如,一个或多个体细胞改变)的数量的函数,其中所述改变的数量排除:
(i)亚基因组间隔(例如,编码亚基因组间隔)中的功能改变;以及
(ii)亚基因组间隔(例如,编码亚基因组间隔)中的种系改变。
用于肿瘤样品的多基因分析的方法的实施方案的流程图描绘提供在图1a-1f中。
本公开包括表5(附录a),所述表5是说明书的一部分并且以引用的方式整体并入本文。
实施例
通过以下实施例进一步说明了本发明,所述实施例不应解释为限制性的。本申请全文中引用的所有参考文献、附图、序列表、专利和公开的专利申请的内容均以引用的方式并入本文。
实施例1:全基因组突变负荷与通过靶向基因测量的突变负荷的比较
在本实施例中,确定了通过靶向315个基因(1.1mb的编码基因组)的全面基因组谱分析(cgp)测试所测量的tmb是否可提供对全外显子组tmb的精确评价。证明了通过靶向的全面基因组谱分析测试得到的对tmb的精确测量。
方法
分析tcga数据
tcga数据获自公共存储库(癌症基因组图谱研究网络(cancergenomeatlasresearchnetwork)等natgenet2013;45:1113-20)。对于这个分析,将通过tcga所确定的所谓体细胞变体用作原始突变计数。38mb用作外显子组大小的估计值。对于下采样分析,使用针对外显子组的范围为每部分0-10mb的各个部分,在全外显子组tmb=100个突变/mb、20个突变/mb以及10个突变/mb下的二项分布来模拟观察到的突变数/mb1000次。黑色素瘤tcga数据获自dbgap登录号phs000452.v1.p1(berger等nature2012;485:502-6)。
肿瘤突变负荷
不希望受理论约束,在本实施例中,如下确定肿瘤突变负荷。肿瘤突变负荷被测量为每兆碱基的检查的基因组,体细胞突变、编码突变、碱基取代突变和插入缺失突变的数量。如下所述,在过滤前初步对靶向基因编码区中的所有碱基取代和插入缺失(包括同义改变)进行计数。对同义突变进行计数以便减少采样噪声。虽然同义突变不可能直接参与产生免疫原性,但它们的存在是突变过程的信号,所述突变过程也引起基因组中其他地方的非同义突变和新抗原。非编码改变不计数在内。作为cosmic中已知的体细胞改变和肿瘤抑制基因中的截短列出的改变不计数在内,因为测试的基因偏向于具有癌症中的功能突变的基因(bamford等brjcancer2004;91:355-8)。通过体细胞-种系-接合性(sgz)算法预测为种系的改变不计数在内(sun等cancerresearch2014;74(19s):1893-1893)。在临床样本组群中反复预测为种系的改变不计数在内。dbsnp中已知的种系改变不计数在内。针对exac数据库中的两个或更多个计数存在的种系改变不计数在内(lek等nature2016;536:285-91)。为了计算每兆碱基的tmb,将计数的突变总数除以靶向区域的编码区的大小。随后使用非参数曼-惠特尼u-检验来测试两个群体之间平均值差的显著性。
结果
执行对公共可用的tcga全外显子组测序数据集(癌症基因组图谱;cancergenome.nih.gov)的初步分析,以确定使用靶向基因(例如,图3a-3b中所示的基因)测量的突变负荷是否将提供对全基因组突变负荷的精确评价。从tcga下载来自35种相异研究/疾病的7,001个样本的完整突变识别数据。对于全外显子组数据集和通过使用图3a-3b中所示的基因的测试靶向的基因中存在的这些突变的数量,对体细胞编码突变的数量进行计数。这些数据提供在表5(附录a)和/或图5-6中示出的散点图中。来自全外显子组的突变负荷仅与来自图3a-3b中所示的基因的突变负荷相关,其中确定系数(r平方)为0.974。
进一步分析包括来自作为癌症基因组图谱的一部分公布,共检查了8,917个癌症样本的35项研究的全外显子组测序数据(癌症基因组图谱研究网络等natgenet2013;45:1113-20)。确定突变的总数量,并且与通过测试靶向的315个基因中的突变数量进行比较。这些结果也同样是高度相关的(r2=0.98)。
这些结果证明,使用靶向数百个基因的整个编码区的cgp(例如,使用仅来自通过测试靶向的基因的数据,所述测试使用图3a-3b中所示的基因),可精确评价全外显子组突变负荷。
总之,本研究表明,使用1.1mb全面基因组谱分析测定计算的肿瘤突变负荷与突变负荷的全外显子组测量一致。这表明靶向数百个基因的整个编码区的cgp覆盖了足够的基因组空间以精确评价全外显子组突变负荷。发现过滤掉种系改变和罕见变体可用于获得tmb的精确测量,并且这可对于在测序数据集中没有很好代表的种族背景的患者尤其有用。这些发现表明cgp是一种精确、成本效益好和临床可用的用于测量tmb的工具。下采样分析的结果表明,当测序1.1mb时由于采样引起的测量变化是可接受的低,引起在tmb水平范围内高度精确的tmb识别。随着测序的mb数量减少,这种采样变化增加,尤其是在较低的tmb水平下。
实施例2:癌症类型的突变负荷景象
在本实施例中,描述了跨不同的≥100,000个癌症样本组群的tmb分布,并且测试了超过100种肿瘤类型的体细胞改变与tmb之间的关联性。发现患者子集在几乎所有癌症疾病类型(包括许多罕见的肿瘤类型)中都表现出高tmb。发现tmb随着年龄而显著增加,在10岁与90岁之间示出2.4倍差异。使用靶向大约1.1mb编码基因组的cgp测定,发现存在许多疾病类型,其中相当一部分具有高tmb的患者可受益于免疫疗法。
本研究基于来自不同类型的>100,000个患者肿瘤的全面基因组谱分析(cgp)的数据,提供对人癌症范围内的tmb景象的更好理解。本实施例中描述的分析根据定量癌症中突变负荷的现有数据而显著扩展,从而为许多先前未描述的癌症类型提供数据。提供新数据以支持可受益于免疫疗法的患者群体的合理扩展,并且允许在未经测试的癌症类型中对免疫疗法药剂的临床试验进行知情设计。
方法
全面基因组谱分析
如先前详细描述,执行cgp(frampton等natbiotech2013;31:1023–1031;he等blood2016;127:3004-14;foundationone测定(cambridge,ma,usa))。简而言之,通过评述苏木精和曙红(h&e)染色的载玻片来确认每个病例的病理诊断,并且所有进展至dna提取的样品都包含最少20%的肿瘤细胞。将来自185、236、315或405个癌症相关基因的外显子区域和来自通常在癌症中重排的19、28或31个基因的选择内含子的杂交捕获应用于从福尔马林固定、石蜡包埋的临床癌症样本中提取的≥50ng的dna。这些文库被测序为高且均匀的中值覆盖度(>500x)并且评价碱基取代、短插入和缺失、拷贝数改变和基因融合/重排(frampton等natbiotech2013;31:1023–1031)。在分析中使用来自三个版本测定中的每一个的数据。
肿瘤突变负荷
不希望受理论约束,在本实施例中,如实施例1中所述确定肿瘤突变负荷。
组群选择
将来自相同患者的重复测定结果从初始的102,292个样品临床组群中排除,并且排除具有小于300x中值外显子覆盖度的样品以制备92,439个样品的分析集合。对于癌症类型的分析,在样品水平过滤后,它们必须包含最少50个独特的样本。
检查在实验室中进行谱分析的患者组群的tmb景象。在为102,292名癌症患者进行常规临床护理过程中执行cgp(参见本实施例的“方法”章节)。独特的患者组群包含41,964名男性患者和50,376名女性患者。样本收集时患者的中值年龄为60岁(范围:<1岁至>89岁),并且2.5%的病例来自18周岁以下的儿科患者。这组数据提供了541种相异癌症类型以用于分析。值得注意的是,大多数样本来自患有显著预治疗、晚期和转移性疾病的患者。在整个数据集中,中值突变负荷为3.6个突变/mb,其中范围为0–1,241个突变/mb。这与先前来自全外显子组研究(alexandrov等nature2013;500:415-21;lawrence等nature2013;499:214-8)的突变负荷估计值一致。发现tmb显著增加与年龄增加相关(p<1x10-16),虽然影响大小很小(图7)。10岁时的中值tmb为1.67个突变/mb,并且88岁时的中值tmb为4.50个突变/mb。对数据的线性模型拟合预测了,10岁与90岁之间tmb的2.4倍差异与这些年龄的中值tmb差异一致。女性患者与男性患者之间的中值突变负荷不存在统计学显著的差异(图8a)。
检查了167种相异癌症类型的tmb,对于所述癌症类型已测试了多于50个样本(图9,表6)。中值tmb范围广泛,从骨髓的骨髓增生异常综合征中的0.8个突变/mb至皮肤鳞状细胞癌中的45.2个突变/mb。发现儿童恶性肿瘤(患者年龄小于18岁)的tmb(中值1.7个突变/mb)低于成人恶性肿瘤(中值3.6个突变/mb)。儿科患者常见的疾病类型诸如白血病、淋巴瘤和神经母细胞瘤具有低tmb,肉瘤也是如此(表6)。
表6.按疾病计的tmb特性总结
*ci:置信区间
已知具有显著诱变剂暴露的疾病诸如肺癌和皮肤癌是更高度突变的(中值tmb分别为7.2个突变/mb和13.5个突变/mb)。目前批准用于免疫疗法的疾病适应症(包括黑色素瘤、非小细胞肺癌(nsclc)和膀胱癌)具有高tmb(参见表6)。鉴定具有高tmb的另外癌症类型可代表扩展积极响应于检查点抑制剂阻断的适应症列表的机会。这些包括皮肤鳞状细胞癌、肺小细胞未分化癌、弥散性大b细胞淋巴瘤,以及许多其他类型的癌症(图6a-6c)。除了鉴定具有高总体tmb的另外癌症类型之外,几乎每种癌症类型都发现具有高tmb的病例(参见表6-7)。这提高了可在几乎每种类型的癌症中鉴定出可受益于免疫疗法的具有高tmb的患者的可能性。例如,在软组织血管肉瘤中,虽然中值突变负荷为3.8个突变/mb,但13.4%的病例具有大于20个突变/mb。总体而言,鉴定了影响8种组织的20种肿瘤类型,其中大于10%的患者具有高tmb,并且鉴定了影响19种组织的38种肿瘤类型,其中大于5%的患者具有高tmb(参见表7)。
表7.大于5%的样本示出高tmb(>20个突变/mb)的疾病适应症。
总之,本研究表征并且提供了大量数据,所述数据描述了来自晚期疾病(包括许多先前未描述的癌症类型)的大于100,000个临床癌症样本的肿瘤突变负荷。这些数据可用于指导更广泛适应症的免疫疗法临床试验的设计。目前,靶向ctla-4、pd-1和pd-l1的免疫疗法被批准用于少数适应症、黑色素瘤、膀胱癌、nsclc和肾细胞癌。据观察,黑色素瘤和nsclc代表了一些最高突变负荷适应症。鉴定了若干种具有高突变负荷的新型疾病类型,所述疾病类型可以是免疫肿瘤学治疗发展的良好靶标。另外,在许多癌症类型中观察到广泛的tmb。发现可存在许多疾病类型,其中相当一部分患者可受益于这些疗法。总体而言,鉴定了影响8种组织的22种肿瘤类型,其中大于10%的患者具有高tmb。
实施例3:全面基因组谱分析以评价肺癌中的突变负荷
肺癌提出了一项管理挑战,特别当egfr、alk或ros1突变无法检测到并且细胞毒性疗法失败时。为了研究突变负荷与新型免疫治疗剂(例如,pd-1/pd-l1和ctla4抑制剂)疗效的关联性,通过在对患有肺癌的患者的临床护理过程中执行的基因组谱分析来评价突变负荷。
方法
简而言之,从来自患有肺癌的患者的40微米ffpe切片中提取dna。针对315个癌症相关的基因加上来自28个频繁在癌症中重排的基因的内含子,在杂交捕获、基于衔接子连接的文库上执行cgp以达到663×的中值覆盖深度。不希望受理论约束,在本实施例中,突变负荷被表征为在过滤以除去如本文所述的已知体细胞改变和功能改变后每兆碱基(mb)的碱基取代或插入缺失的数量,假定这些是用杂交捕获选择的。
ffpe肿瘤样品
样品要求如下:表面积:≥25mm2;样品体积:≥1mm3;有核细胞结构:≥80%或≥30,000个细胞;肿瘤含量:≥20%;组织不足以分析的患者的分数:10%-15%。
测序文库制备
实验室方法需要≥50ng的dsdna(由picogreen定量)。通过超声(covaris)将dna片段化并且用于“利用磁珠”的文库构建。通过与生物素化的dna寡核苷酸杂交来捕获dna片段。在illuminahiseq平台上执行49×49配对末端测序以达到>500×平均独特覆盖度,其中在>99%的外显子处>100×。
分析流程
通过贝叶斯算法来分析碱基取代。通过局部组装来评估短插入/缺失。通过与方法匹配的正常对照比较来分析拷贝数改变。通过分析嵌合读取对来检查基因融合。
分析方法对在任何突变等位基因频率下存在的变体具有灵敏性,并且能够使用基于debruijn图的局部组装来检测长(1-40bp)插入缺失变体。分析方法还使用比较基因组杂交(cgh)样的读取深度分析,以用于评价拷贝数改变(cna)。
临床报告
报告方法提供解释而不需要匹配的正常值。除去来自千人基因组计划的种系变体(dbsnp135)。已知的司机改变(cosmicv62)被强调为生物学显著的。为每个改变都提供了生物医学文献和目前临床试验的简明摘要。
突变负荷分析方法
突变负荷算法的目标是定量在
计数在foundationone测试上检测到的所有短变体改变(碱基取代和插入缺失)。计数所有编码改变,包括沉默改变。非编码改变不计数在内。具有已知功能状态(作为cosmic数据库中的已知体细胞改变存在的;cancer.sanger.ac.uk/cosmic)和可能的功能状态(肿瘤抑制基因中的截短)的改变不计数在内。dbsnp数据库(www.ncbi.nlm.nih.gov/snp)中的已知种系改变不计数在内。针对exac数据库(exac.broadinstitute.org)中的两个或更多个计数存在的种系改变不计数在内。通过体细胞-种系-接合性(sgz)算法(例如,在国际申请公布wo2014/183078、美国申请公布2014/0336996以及sun等cancerresearch2014;74(19s):1893-1893中所述),在被评价的样品中预测为种系的改变不计数在内。通过sgz算法,在>60,000个临床样本的组群中以高置信度预测为种系的改变不计数在内。为了计算每兆碱基的突变负荷,将计数的突变总数除以测试的编码区靶区域,所述编码区靶区域对于目前的测试版本为1.252兆碱基。
结果
评价了来自总共10,676例肺腺癌、1,960例肺鳞状细胞癌、220例肺大细胞癌和784例肺小细胞癌的基因组谱分析。肺癌患者的中值年龄为66周岁,其中男:女比率为0.9:1。每兆碱基的平均突变被评价为0至984的范围,并且25%阈值、中值阈值和75%阈值为2.7、7.2和22.5。
肺癌患者组群的临床特征在表8中示出。肺癌的突变负荷特征在表9中示出。
表8.肺癌患者组群的临床特征
表9.肺癌的突变负荷特征
临床组群中的突变负荷分布在图7a-7d中示出。肺癌中的突变流行率在图8a-8e中示出。
总之,在患有肺癌的患者中可看到高度可变的突变负荷。当患者匹配的正常样本不可用时,通过计算而精确区分体细胞突变与正常突变的能力是必不可少的。相当一部分肺癌病例具有高突变负荷(39%≥10个/mb;13%≥20个/mb),并且是免疫治疗剂临床试验的潜在候选者。
实施例4:全面基因组谱分析以评价结肠直肠腺癌中的突变负荷
结肠直肠腺癌仍然是一项临床挑战,特别当kras或nras基因突变并且细胞毒性疗法失败时。为了研究肿瘤突变负荷与来自免疫检查点抑制剂的预测益处的关联性,在使用基因组谱分析的常规临床护理过程中评价结肠直肠腺癌样品中突变负荷与临床相关基因组改变之间的关系。
方法
从来自患有结肠直肠腺癌的患者的40微米ffpe切片中提取dna。针对315个癌症相关的基因加上来自28个频繁在癌症中重排的基因的内含子,在杂交捕获、基于衔接子连接的文库上执行cgp以达到698×的平均覆盖深度。不希望受理论约束,在本实施例中,突变负荷被表征为在过滤以除去如本文所述的已知体细胞改变和功能改变后每兆碱基(mb)的碱基取代或插入缺失的数量,假定这些是用杂交捕获选择的。
样品要求、测序文库制备、分析流程、临床报告和突变负荷分析方法如实施例3中所述。
结果
评价了来自总共6,742例结肠腺癌和1,176例直肠腺癌的基因组谱。结肠直肠腺癌患者的中值年龄为57周岁,其中男:女比率为1.2:1。每兆碱基的平均突变被评价为0至866的范围,并且25%阈值、中值阈值和75%阈值为2.7、4.5和6.3。
在174(2.2%)例、191(2.4%)例、315(3.9%)例或283(3.6%)例结肠直肠腺癌中检测到错配修复基因mlh1、msh2、msh6或dna聚合酶基因pold1中的遗传改变,这分别为30、23、29或15的中值肿瘤突变负荷相关。然而,在这个组群中十个最频繁改变的基因–apc(76%)、tp53(76%)、kras(51%)、pik3ca(18%)、smad4(15%)、fbxw7(10%)、sox9(10%)、myc(8%)、braf(8%)和pten(8%)–与肿瘤突变负荷的差异无关。
结肠直肠腺癌患者组群的临床特征在表10中示出。结肠直肠腺癌的突变负荷特征描述于表11中。
表10.结肠直肠腺癌患者组群的临床特征
表11.结肠直肠腺癌的突变负荷特征
临床组群中的突变负荷分布在图9a-9b中示出。结肠直肠腺癌的突变流行率在图10a-10c中示出。
总之,在临床护理过程中的cgp可用于评价结肠直肠腺癌中的突变负荷。dna错配修复基因中的突变与预期的更高突变负荷相关。相当一部分结肠直肠腺癌病例具有高突变负荷(9%≥10个/mb;5%≥20个/mb),并且是免疫治疗剂临床试验的潜在候选者。需要将cgp并入正在进行的前瞻性免疫疗法试验和临床实践中,以改进这些关系。
实施例5:全面基因组谱分析以评价二十四种类型人肿瘤中的突变负荷
为了研究肿瘤突变负荷与来自免疫检查点抑制剂的预期益处的关联性,在使用基因组谱分析的常规临床护理过程中评价24种类型肿瘤中突变负荷的分布。
方法
从来自患有二十四种类型肿瘤中的一种的患者的40微米ffpe切片中提取dna。针对315个癌症相关的基因加上来自28个频繁在癌症中重排的基因的内含子,在杂交捕获、基于衔接子连接的文库上执行cgp以达到大于500×的平均覆盖深度。不希望受理论约束,在本实施例中,突变负荷被表征为在过滤以除去如本文所述的已知体细胞改变和功能改变后每兆碱基(mb)的碱基取代或插入缺失的数量,假定这些是用杂交捕获选择的。
样品要求、测序文库制备、分析流程、临床报告和突变负荷分析方法如实施例3中所述。
结果
评价了来自总共15,508个肿瘤样本的基因组谱患者组群的中值年龄为60周岁,其中男:女比率为0.6:1。每兆碱基的平均突变被评价为0至689的范围,并且25%阈值、中值阈值和75%阈值为1.8、3.6和5.4。
患者组群的临床特征在表12中示出。二十四种类型肿瘤的突变负荷特征描述于表13中。24种不同肿瘤中的tmb分布在图11中示出。
表12.癌症患者组群的临床特征
表13.二十四种类型肿瘤的突变负荷特征
与本文描述的方法和系统相关的另外实施例描述于例如国际申请公布no.wo2012/092426的实施例1-17、国际申请公布no.wo2016/090273的实施例16和17中,前述公布和实施例的内容以引用的方式整体并入。
以引用的方式并入
本文提到的所有出版物、专利和专利申请以引用的方式整体并入本文,如同每个单独的出版物、专利或专利申请具体地和单独地表示为以引用的方式并入。在发生冲突的情况下,本申请(包括本文中的任何定义)将占据主导地位。
还以引用的方式整体并入的是任何多核苷酸和多肽序列,所述多核苷酸和多肽序列参考与公共数据库中的条目相关的登录号,诸如由万维网tigr.org上的基因组研究所(instituteforgenomicresearch,tigr)和/或万维网ncbi.nlm.nih.gov上的美国国家生物技术信息中心(nationalcenterforbiotechnologyinformation,ncbi)维护的那些。
等效方案
本领域技术人员仅仅使用常规实验将认识到或能够确定本文描述的本发明的具体实施方案的许多等效方案。此类等效方案意图涵盖于以下权利要求书中。
表5(附录a)
表5(附录a)
表5(附录a)
表5(附录a)
表5(附录a)
表5(附录a)
表5(附录a)
表5(附录a)
表5(附录a)
表5(附录a)
表5(附录a)
表5(附录a)
表5(附录a)
表5(附录a)
表5(附录a)
表5(附录a)
表5(附录a)
表5(附录a)
表5(附录a)
表5(附录a)
表5(附录a)
表5(附录a)
表5(附录a)
表5(附录a)
表5(附录a)
表5(附录a)
表5(附录a)
表5(附录a)
表5(附录a)
表5(附录a)
表5(附录a)
表5(附录a)
表5(附录a)
表5(附录a)
- 还没有人留言评论。精彩留言会获得点赞!