细叶石仙桃药材指纹图谱的建立方法及其指纹图谱与流程
本发明涉及中药材的指纹图谱技术领域,具体涉及一种细叶石仙桃药材指纹图谱的建立方法及其指纹图谱。
背景技术:
中药虽然已经有几千年的应用发展历史,但如何有效的评价中药材质量的好坏,一直是中药材研究与应用的重点和难点。随着科技的发展,指纹图谱质控技术在中药领域被提出并得到广泛认可,其在中药材质控中的应用也越来越多;中药材指纹图谱是指中药材经过适当处理后,采用一定的分析手段和检测仪器得到的,能够标示该中药材特性的图谱,它是现阶段可以较全面地反映中药材内在质量的最有效的手段之一,也得到了国际社会的认可。
细叶石仙桃pholidotacantonensisrolfe.是一种常用中药,为兰科石仙桃属植物,常以假鳞茎或全草入药,又名双叶岩珠等,俗称石穿盘(广西)、浮石斛等,产于中国浙江、江西、福建、台湾、湖南、广东和广西。味微苦、甘,性凉,归肺经、肾经,具有滋阴润肺、清热凉血、解毒、消瘀等功效。用于咳嗽,吐血,痢疾,白带,疳积。人们对细叶石仙桃开展的化学成分研究中发现,细叶石仙桃含黄酮类、菲类、酚类、萜类、甾体类、多糖、脂肪族类及矿物质等化学成分,具有麻醉、镇痛、抑制中枢神经系统、抗疲劳、耐缺氧等药理作用。目前国内外对细叶石仙桃的研究主要集中在化学成分的提取、分离、分析和抑菌及中药材真伪鉴别等方面的研究,而在hplc指纹图谱方面的研究少有报道。
而由于不同的气候条件等外界环境因素,各产地的细叶石仙桃样品中所含成分有一定差别。但现有技术中对细叶石仙桃的检测大都只能粗略地鉴别细叶石仙桃的真伪,不能全面、客观的检测和评价细叶石仙桃的质量。因此为全面、准确、有效地控制细叶石仙桃的质量及合理利用细叶石仙桃,有必要建立细叶石仙桃的指纹图谱,弥补细叶石仙桃质量控制不足,使其更加完善、科学。
技术实现要素:
本发明的目的是提供一种细叶石仙桃药材(pholidotacantonensisrolfe.)指纹图谱的建立方法及其指纹图谱,利用该图谱能够明显区别细叶石仙桃和其他药材,并能区别不同产地的细叶石仙桃,监控细叶石仙桃药材的质量及鉴别真伪,确保药材的真实、安全、有效、稳定和一致性,为进一步开发、制定其中药材标
准提供参考依据,规范、保障临床用药。
为实现上述目的,本发明采用如下技术方案:
一种细叶石仙桃药材指纹图谱的建立方法,包括以下步骤:
(1)对照品溶液的制备:精密称取没食子酸标准品、绿原酸标准品、丁香醛标准品,分别置10ml容量瓶中,用甲醇溶解并定容至刻度,摇匀,制得浓度分别为0.125g/ml、0.0588g/ml、0.112g/ml的溶液,再用0.45μm微孔滤膜过滤,即为对照品溶液;
(2)供试品溶液的制备:取细叶石仙桃粗粉2.5g,精密称定,置具塞锥形瓶中,加80ml体积浓度为80%的甲醇超声处理1.5h,用体积浓度为80%的甲醇溶液补重,摇匀后过滤,取续滤液,离心10min,取上清液,用0.45μm微孔滤膜过滤,即为供试品溶液;
(3)高效液相色谱分析
色谱条件为:用agilent5tc-c18,250mm×4.6mm,5μm色谱柱;以乙腈为流动相b,以体积百分含量为0.4%的磷酸溶液为流动相d,采用以下梯度洗脱的方式进行梯度洗脱:从0~26mim,流动相b的体积百分含量由18%提高至33%,流动相d的体积百分含量由82%降低至67%;26~38mim,流动相b的体积百分含量由33%提高至38%,流动相d的体积百分含量由67%降低至62%;38~58mim,流动相b的体积百分含量由38%提高至50%,流动相d的体积百分含量由62%降低至50%;58~68mim,流动相b的体积百分含量由50%提高至60%,流动相d的体积百分含量由50%降低至40%;流速:1.0ml·min-1;进样量:10μl;检测波长为280nm,柱温为30℃;
(4)测定:分别精密吸取对照品溶液与供试品溶液各10μl,注入高效液相色谱仪,测定,记录60分钟色谱图,以绿原酸为特征图谱参照物,以绿原酸色谱峰为参照物峰,测定10批细叶石仙桃药材的指纹图谱,得到由其共有特征峰构成的细叶石仙桃药材的标准指纹图谱,以确定的参照物色谱峰的保留时间为基准,计算其它共有色谱峰的相对保留时间,所述标准指纹图谱中,共有峰有7个,其中2号峰为绿原酸。
本发明的另一目的是提供上述细叶石仙桃药材指纹图谱的建立方法构建得到的细叶石仙桃药材的标准指纹图谱,所述的细叶石仙桃药材的标准指纹图谱具有7个共有特征峰,以参照物峰相应的峰为s峰,所述7个共有特征峰的相对保留时间如下:峰1:2.973,峰2:3.832,峰3:9.542,峰4:11.35,峰5:16.907,峰6:26.71,峰7:28.748。
本发明的有益效果为:
本发明一种细叶石仙桃药材指纹图谱的建立方法及其指纹图谱,具有重现性好、精密度高、稳定性好的特点,通过该方法构建得到细叶石仙桃药材的标准指纹图谱,通过比较指纹图谱中共有峰的有无,能有效地鉴别细叶石仙桃药材的真伪及产地,用于反映细叶石仙桃药材质量的优劣,完善细叶石仙桃药材的质量评价体系。通过细叶石仙桃药材的标准指纹图谱,可较全面地对细叶石仙桃药材的内在质量进行控制与评价,监控细叶石仙桃药材的质量及鉴别真伪,确保药材的真实、安全、有效、稳定和一致性,为进一步开发、制定其中药材标准提供参考依据,规范、保障临床用药。
附图说明
图1为色谱柱agilent5tc-c18(2)(250mm×4.6mm,5μm)的hplc色谱图;
图2为色谱柱agilentzorbaxsb-c18(250mm×4.6mm,5μm)的hplc色谱图;
图3为流动相乙腈-水系统色谱图;
图4为流动相乙腈-0.1%磷酸系统色谱图;
图5为流动相乙腈-0.4%磷酸系统色谱图;
图6为流动相甲醇-水系统色谱图;
图7为流动相甲醇-0.1%磷酸系统色谱图;
图8为流动相甲醇-0.2%磷酸系统色谱图;
图9为流速为1.0ml/min的色谱图;
图10为流速为0.8ml/min的色谱图;
图11为流速为0.5ml/min的色谱图;
图12为超声提取的色谱图;
图13为回流提取的色谱图;
图14为热浸12h提取的色谱图;
图15为冷浸24h提取的色谱图;
图16为空白溶剂试验hplc色谱图;
图17为延长冲洗试验hplc色谱图;
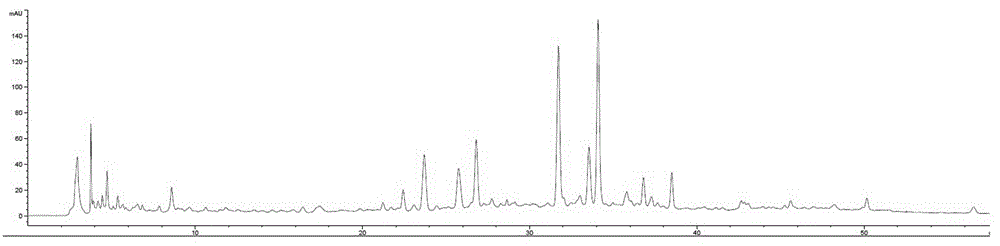
图18为细叶石仙桃药材的标准指纹图谱;
图19为10批细叶石仙桃药材hplc图谱叠加图;
图20为聚类分析结果图;
图21为没食子酸色谱图;
图22为绿原酸色谱图;
图23为丁香醛色谱图。
具体实施方式
以下结合具体实施例对本发明进行进一步详细的叙述。
实施例1
一种细叶石仙桃药材指纹图谱的建立方法,具体实验步骤如下:
一、试验材料
1.1、实验材料
10批次细叶石仙桃药材样品主要采集于广西不同产地,详见(表1),经广西中医药大学滕建北教授鉴定为兰科石仙桃属植物细叶石仙桃pholidotacantonensisrolfe.全草。
1.2、仪器
美国agilent1260高效液相色谱仪(g1311c四元泵,g1329b自动进样器,g1316a柱温箱和g1314b检测器);kq-500da数控超声波清洗器(昆山市超声仪器有限公司);bsa224s电子分析天平(赛多利斯科学仪器(北京)有限公司);
电热恒温鼓风干燥箱(上海一恒仪器有限公司);高速万能粉碎机(湖南湘仪实验仪器开发有限公司);hws-28数显恒温水浴锅(上海齐欣科学仪器有限公司);
h1650-w型高速台式离心机(湖南湘仪实验仪器开发有限公司);shb-iiia循环水式多用真空泵(郑州长城科工贸有限公司);milli-qadvantagea10超纯水系统(美国默克密理博公司)。
1.3试剂
甲醇(美国fisher公司,色谱纯);乙腈(美国fisher公司,色谱纯);甲醇、95%乙醇、无水乙醇、石油醚、乙酸乙酯(国药集团化学试剂有限公司,分析纯);磷酸(天津市科密欧化学试剂有限公司,色谱纯);绿原酸(中国食品药品检定研究院,批号110753-201415,供含量测定使用);没食子酸(中国食品药品检定研究院,批号110831-201605,供含量测定使用);丁香醛(中国食品药品检定研究院,批号110623-201704,供含量测定使用);水为超纯水。
二、实验方法
按照如下方法建立细叶石仙桃药材指纹图谱:
2.1色谱柱的选择
不同来源、品牌的色谱柱具有一定的差异,同一来源的色谱柱也有不同程度的差
别,柱效(理论塔板数)、保留时间、分离度、对称因子都有差别,在本实验中色谱条件相同的情况下,进行了不同厂家同一规格的c18两根高效液相色谱柱进行考察,结果色谱图如下见图1-图2。
色谱柱规格如下:
(1)agilent5tc-c18(2)(250mm×4.6mm,5μm)批号518925-902。
(2)agilentzorbaxsb-c18(250mm×4.6mm,5μm)批号880975-902。
实验结果表明,在agilent5tc-c18(2)柱(250mm×4.6mm,5μm)色谱柱中,各峰的分离度较好、基线较平稳。
2.2流动相系统的选择
本试验考察了6种不同的流动相:乙腈-水、乙腈-0.1%磷酸、乙腈-0.4%磷酸、甲醇-水、甲醇-0.1%磷酸、甲醇-0.4%磷酸,流动相的分离效果,其色谱图如图3-图8所示。
2.3检测波长的选择
为选得细叶石仙桃药材最佳的吸收波长,本试验采用二极管阵列检测器对样品在
波长为265nm、270nm、275nm、280nm、285nm、290nm、295nm、315nm下的色谱
情况进行考察,通过对比得出在波长为280nm时所得到的色谱峰的分离度、峰面积、保留时间、对称因子比较好及得到峰的个数比较多,基线较平稳,峰高较均匀,故本研究选用在280nm波长下测定。
2.4柱温的选择
恒定的柱温是影响指纹图谱技术的关键之一,在不同的柱温情况下,各组分出峰
的时间可能不太相同,对分离效果也会有一定的影响,本试验考察了20℃、25℃、30℃三个不同的柱温进行比较,在其它色谱条件相同情况下,30℃时各峰分离效果最好,所以最后确定柱温为30℃。
2.5流速的选择
流速对色谱的检测结果也有一定的影响,它是调整分离度和出峰时间的重要可选
择参数。本试验考察了不同流速(1ml/min、0.8ml/min、0.5ml/min)对细叶石仙桃色谱峰的分离效果、保留时间、峰面积、对称因子的影响,结果显示流速为1ml/min时得到的图谱分离度比较好(见图9-图11),故本研究选择采用的流速为1ml/min。
2.6进样量的确定
进样量对实验的检测结果和仪器也会产生一定的影响。进样量过少则检测不出或
者检测不完全;进样量高可以降低基线漂移程度,但进样量过高则会污染色谱槽。本研究考察了12μl、10μl、8μl的进样量,结果显示10μl的进样量的分离度相对较好,故选择的进样量为10μl。
2.7确定指纹图谱色谱条件
根据以上考察的结果,液相的色谱条件确定为agilent5tc-c18(2)(250mm×4.6mm,5μm);流动相:乙腈-0.4%磷酸,;检测波长为:280nm;流速:
1.0ml·min-1;柱温:30℃;进样量:10μl;洗脱程序见表2。
三、制备方法及方法学考察
3.1供试品制备方法的确定
细叶石仙桃的主要化学成分有黄酮类、酚类、萜类、甾体、多糖等,不同成分的
极性不同,故需要考察不同的提取溶液、提取方法及料液比,尽可能提取出更多的成分,建立较全面的指纹图谱,更准确反映出细叶石仙桃化学成分的整体情况。
3.1.1供试品提取溶剂的选择
不同的提取溶剂极性的不同,往往会产生不同的提取效果,故提取溶剂对于研究
样品中不同化学成分的提取、分离有着重要的影响。本研究考察了水、无水乙醇、乙酸乙酯、石油醚、90%甲醇、80%甲醇、70%甲醇、60%甲醇对细叶石仙桃中化学成分提取的效果,根据得到的色谱峰,通过直观的指纹图谱对比,并结合分离度、对称因子等相关指标分析,最终采用浓度为80%的甲醇作为本次究的提取溶剂。
3.1.2提取方法的选择
不同方式的提取方法对样品中的化学成分的提取效果也有重要的影响,本研究考
察了超声、热回流/热浸、冷浸提取四种方法,具体结果如图12-15,直观的图谱比较四种提取方法的峰形及出峰时间基本相同,结合分离度及方法、时间,最终采用超声法提取。同时考察了提取时间,根据谱图结果,综合考虑各方面因素最终采用超声提取1.5h。
3.1.3料液比的选择
不同的料液比对研究样品中的化学成分的提取效果同样也有重要的影响,本研究
考察了1.5g/80ml、2g/80ml、2.5g/80ml、3g/80ml四种提取药量,得到色谱图,直观的图谱对比四种提取结果的峰形图,其出峰时间基本相同,结合分离度等因素,最终采用的料液比为1:32。
综上,最终择优采用的供试品溶液制备方法是超声法提取1.5h、提取溶剂为80%甲醇,料液比1:32。
3.1.4供试品溶液的制备
取细叶石仙桃粗粉约2.5g,精密称定,置具塞锥形瓶中,加80ml80%甲醇超声
1.5h,用80%甲醇溶液补重,摇匀后过滤,取续滤液,离心10min,取上清液,用0.45μm微孔滤膜过滤,即为供试品溶液。
3.2对照品溶液的制备
精密称取没食子酸标准品、绿原酸标准品、丁香醛标准品,分别置10ml容量瓶
中,用甲醇溶解并定容至刻度,摇匀,制得的浓度分别为0.125g/ml、0.0588g/ml、
0.112g/ml,再用0.45μm微孔滤膜过滤,即为对照品溶液。
3.3指纹图谱的方法学考察
3.3.1精密度试验
取桂林荔浦东昌镇产细叶石仙桃药材的干燥粉末,用分析天平进行精密称定
2.5g,按照3.1.4项供试品溶液制备的方法,制备同一供试品,按2.7项下色谱条件,连续进样6次,记录图谱,各共有峰保留时间rsd值在0.0352%~0.3091%之间,各色谱峰面积rsd值在0.1914%~1.7371%之间,表明本研究所用的仪器精密度良好,结果见表3-表4。
3.3.2重复性试验
取6份桂林荔浦东昌镇产细叶石仙桃药材粉末各约2.5g,精密称定,按3.1.4项下供试品制备方法制备,在2.7项下色谱条件进行测定6次,记录色谱图,考察实验方法的重复性。结果表明,6份样品中各色谱峰的相对保留时间rsd值在0.0601%~0.3443%之间,各色谱峰相对峰面积rsd值在1.5506%~3.7684%之间,表明实验重复性良好。结果见表5-表6。
3.3.3稳定性试验
取桂林荔浦东昌镇产细叶石仙桃药材粉末各约2.5g,精密称定,按3.1.4项下供试品制备方法制备,按2.7色谱条件分别在0h、2h、4h、8h、12h、24h各测定1次,考察样品溶液的稳定性。结果表明,各色谱峰的相对保留时间rsd值在0.1024%~0.4586%之间,各色谱峰的相对峰面积rsd值在0.7196%~2.4712%之间,表明供试品25h内稳定性良好。结果见表7-表8。
四、广西细叶石仙桃指纹图谱的建立与评价
4.1空白试验
为了排除其他干扰成分及溶剂峰的存在对实验结果的误判,本研究进行了空白试验,实验结果如图16,由此可看出本研究不受溶剂峰干扰。
4.2延长冲洗试验
按2.7项下色谱条件,取供试品溶液10μl进样,进行梯度洗脱130min,在60min
后无其它色谱峰出现,故采集时间为60min(见图17)。
4.3各产地细叶石仙桃药材的指纹图谱采集
取10批细叶石仙桃药材样品,按3.1.4方法制备供试品溶液,分别进样,用2.7色谱条件进行测定,记录10批次细叶石仙桃药材hplc色谱图,得到各批次样品的图谱。从所得图谱中可看出2号峰分离度较好,出峰时间稳定,各产地含量相差不大,且绿原酸是细叶石仙桃的主要成分之一,故选2号峰为细叶石仙桃主要指标成分,将其作为参比峰,标记为s。
4.4指纹图谱的建立
根据《中药注射剂指纹图谱研究的技术要求(暂行)》规定,必须根据10批或
10批以上供试品的检测结果,采用相对保留时间标定中药材的共有指纹峰。使用《中药色谱指纹图谱相似度评价系统2004a版》计算机软件对实验数据进行分析评价。以s3为参照,采用中位数相关系数法对各指纹图谱的色谱峰进行了多点校正和自动匹配,生成对照标准指纹图谱r见图18,并建立10批细叶石仙桃指纹图谱,色谱叠加见图19。(没食子酸色谱图见图21;绿原酸色谱图见图22;丁香醛色谱图见图23。)
4.5共有峰的标定
使用《中药色谱指纹图谱相似度评价系统2004a版》计算机软件对实验数据进
行自动匹配,得到了7个共有峰。其中2号峰为绿原酸,以参照物峰(即绿原酸色谱峰)相应的峰为s峰,所述7个共有特征峰的相对保留时间如下:c4.6广西细叶石仙桃叶药材相似度评价
根据国家药典委员会提供的中药色谱指纹图谱相似度评价系统(2012版)软件,
进行指纹图谱相似度计算,以中位数为相关系数,时间宽度设定为0.1min,通过系统自动匹配,得到了10批广西细叶石仙桃药材高效液相色谱指纹图谱间的相似度,结果10批广西细叶石仙桃与对照标准指纹图谱的相似度值比较均接近0.9,与对照指纹图谱相似性良好。结果见表9。
4.7聚类分析
将10批药材的各共有峰的峰面积导入spss23.0(ibmspssstatistics23.0)软件,以组间链接、欧式距离分类进行系统聚类,分析结果如图20,当距离为15时s1、s2、s7、s8为一类,s3、s4、s5、s9、s10是一类,表明不同批次的药材由于地理位置等环境的影响,所含化学成分有一定的差异。
5.小结
通过本研究采用agilent1260高效液相色谱仪测定,料液比为1:32,80%甲醇超声1.5h,以agilent5tc-c18(2)为色谱柱,柱温30℃,进样量为10μl,以乙腈-0.4%磷酸为流动相进行梯度洗脱,检测时间为60min,在280nm波长下以绿原酸为参照物的检测方法所得到的细叶石仙桃化学成分的特征性指纹图谱,能够明显区别细叶石仙桃和其他药材,并能区别不同产地的细叶石仙桃,该方法可作为细叶石仙桃药材真伪鉴别的有效方法。利用标准指纹图谱能够监控细叶石仙桃药材的质量及鉴别真伪,确保药材的真实、安全、有效、稳定和一致性,为进一步开发、制定其中药材标准提供参考依据,规范、保障临床用药。
实施例2
一种细叶石仙桃药材指纹图谱的建立方法,包括以下步骤:
(1)对照品溶液的制备:精密称取没食子酸标准品、绿原酸标准品、丁香醛标准品,分别置10ml容量瓶中,用甲醇(分析纯)溶解并定容至刻度,摇匀,制得浓度分别为0.125g/ml、0.0588g/ml、0.112g/ml的溶液,再用0.45μm微孔滤膜过滤,即为对照品溶液;
(2)供试品溶液的制备:取细叶石仙桃粗粉2.5g,精密称定,置具塞锥形瓶中,加80ml体积浓度为80%的甲醇超声处理1.5h,用体积浓度为80%的甲醇溶液补重,摇匀后过滤,取续滤液,离心10min,取上清液,用0.45μm微孔滤膜过滤,即为供试品溶液;
(3)高效液相色谱分析
色谱条件为:用agilent5tc-c18,250mm×4.6mm,5μm色谱柱;以乙腈为流动相b,以体积百分含量为0.4%的磷酸溶液为流动相d,采用以下梯度洗脱的方式进行梯度洗脱:从0~26mim,流动相b的体积百分含量由18%提高至33%,流动相d的体积百分含量由82%降低至67%;26~38mim,流动相b的体积百分含量由33%提高至38%,流动相d的体积百分含量由67%降低至62%;38~58mim,流动相b的体积百分含量由38%提高至50%,流动相d的体积百分含量由62%降低至50%;58~68mim,流动相b的体积百分含量由50%提高至60%,流动相d的体积百分含量由50%降低至40%;流速:1.0ml·min-1;进样量:10μl;检测波长为280nm,柱温为30℃;
(4)测定:分别精密吸取对照品溶液与供试品溶液各10μl,注入高效液相色谱仪,测定,记录60分钟色谱图,以绿原酸为特征图谱参照物,以绿原酸色谱峰为参照物峰,测定10批细叶石仙桃药材的指纹图谱,得到由其共有特征峰构成的细叶石仙桃药材的标准指纹图谱,以确定的参照物色谱峰的保留时间为基准,计算其它共有色谱峰的相对保留时间,所述标准指纹图谱中,共有峰有7个,其中2号峰为绿原酸,以参照物峰(即绿原酸色谱峰)相应的峰为s峰,所述7个共有特征峰的相对保留时间如下:峰1:2.973,峰2:3.832,峰3:9.542,峰4:11.35,峰5:16.907,峰6:26.71,峰7:28.748。
- 还没有人留言评论。精彩留言会获得点赞!