一种茯苓质量的检测方法及其指纹图谱的构建方法与流程
本发明涉及质量分析领域,具体涉及一种茯苓质量的检测方法及其指纹图谱的构建方法。
背景技术:
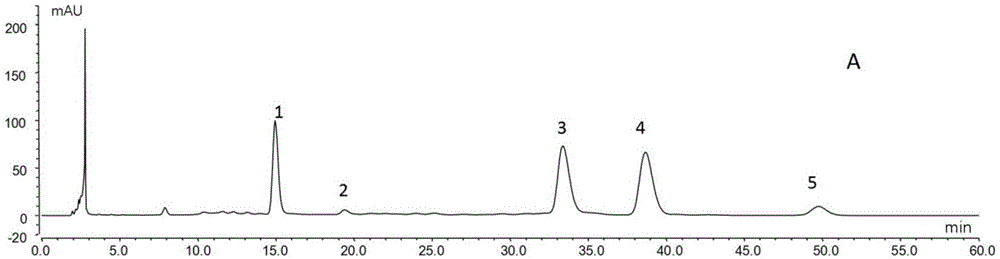
:茯苓为多孔菌科真菌茯苓poriacocos(schw.)wolf的干燥菌核,其味甘、淡,性平,具有利水渗湿、健脾宁心之功效,应用历史悠久且在中医临床有“十药九茯苓”之说,同时其作为传统药食同源物品在保健功能食品也有广泛应用。对于茯苓的质量评价,一直缺乏有效的质量分析方法,至2015年版《中国药典》一部“茯苓”项下仍未建立相应的生物活性物质检测项。已有研究显示茯苓中含有多糖、三萜、甾体、蛋白质、矿物质等化学成分,现一般认为多糖及三萜类是其主要和重要的生物活性物质。针对茯苓中三萜类成分的分析检测方法,已有较多研究,如“波长转换rp-hplc法同时测定茯苓不同部位中5种三帖酸含量”(车爽等,药学学报,2010,45(4):494~497)、“20个产地茯苓三萜成分rp-hplc-elsd指纹图谱”(昝俊峰等,中国实验方剂学杂志,2013年,19(21):65~68)等,但关键问题是茯苓药材中三萜类成分含量极低,较高含量也不过万分之几,对hplc(高效液相色谱法)检测器的灵敏度要求较高,且中医药方剂多采用水煎煮法,而在茯苓的水煎液及其配方颗粒中几乎检测不到茯苓药材中所含三萜酸类化学成分,难以纳入质量检测方法。针对茯苓多糖的分析方法,目前多采用苯酚-浓硫酸显色分光光度法测定,方法的专属性不佳,难以作为有效的质量控制方法。吴建元等人用pmp(1-苯基-3-甲基-5-吡唑啉酮)柱前衍生-hplc的方法检测了茯苓的碱溶性多糖,发现茯苓碱溶性多糖为均多糖,只由一种单糖,即葡萄糖组成,不具有特征性,不能表征茯苓的特征(吴建元等,医药导报,2009,28(9):1213~1214)。现有研究中为了尽可能多的提取出中药药材的更多化学成分,进行质量控制时常采用有机溶剂提取的方式。由于水煎煮法得到的成分含量少、多糖收率较低(例如参见茯苓中多糖的提取及含量测定,周燕霞等,天然产物研究与开发,2003,15(4):330~333),现有技术中对茯苓的水溶性成分研究较少,用水溶性成分对茯苓进行质量评价的文献迄今未检索到。技术实现要素:本发明所要解决的技术问题是为了克服现有技术中缺乏有效的针对茯苓质量的检测方法,提供了一种茯苓指纹图谱的构建方法、一种茯苓质量的检测方法、一种检测茯苓多糖的单糖组成的方法以及一种茯苓多糖提取物的分离方法。本发明所述茯苓质量的检测方法应用了能反映茯苓多糖的单糖组成特征的茯苓指纹图谱,可有效地检测分析茯苓汤剂及其配方颗粒的质量。本发明所述方法操作简单、稳定、重现性好、检测效率高,具有较强实用性。本发明收集不同产地二十余批次的茯苓药材,采用水煎煮法对其水溶性多糖逐一进行单糖组成的研究,通过单糖的衍生化提高hplc的检测灵敏度,并且进行大量实验摸索出了本发明所述的hplc的流动相比例,较好实现对茯苓的单糖组成的分析。为解决上述技术问题,本发明旨在提供一种茯苓指纹图谱的构建方法所述构建方法包括以下步骤:(1)利用高效液相色谱检测茯苓的供试品溶液,(2)将检测结果生成指纹图谱;所述高效液相色谱的色谱柱为反相柱;所述高效液相色谱为使用流动相的体积比为15:85至20:80的乙腈-磷酸盐缓冲溶液进行等度洗脱;所述供试品溶液的制备包括:通过水煎煮法制得茯苓水溶性粗多糖;将所得粗多糖经酸水解后,以1-苯基-3-甲基-5-吡唑啉酮进行衍生化处理,萃取后取水层定容作为供试品溶液。较佳地,所述反相柱为硅胶键合相色谱柱,优选十八烷基硅烷键合硅胶色谱柱或辛烷基硅烷键合硅胶色谱柱。较佳地,所述磷酸盐缓冲溶液的ph值为6.5~7.0,优选6.80。较佳地,所述磷酸盐缓冲溶液的浓度为30~50mmol/l,优选50mmol/l。较佳地,所述磷酸盐缓冲溶液为kh2po4-naoh缓冲溶液。较佳地,所述流动相的流速为0.8~1.2ml/min,优选1ml/min。较佳地,所述反相柱的柱温为25~30℃,优选30℃。较佳地,所述流动相的体积比为18:82或17.5:82.5。较佳地,所述高效液相色谱的检测波长为240~250nm,优选250nm。较佳地,所述高效液相色谱的进样体积为10~20μl,优选10μl。较佳地,所述十八烷基硅烷键合硅胶色谱柱的柱长为150~250mm,优选250mm;和/或,所述十八烷基硅烷键合硅胶色谱柱的内径为2.1~4.6mm,优选4.6mm;和/或,所述十八烷基硅烷键合硅胶色谱柱的填料粒径为3~5μm,优选5μm;更佳地,所述十八烷基硅烷键合硅胶色谱柱为agilentextendc18,4.6*250mm,5μm。较佳地,所述水煎煮法包括以下步骤:(1)将茯苓通过水煎煮制得样品,(2)将(1)中所得样品进行后处理;所述后处理通过以下方法之一进行:方法一为将所述样品经浓缩或水溶,和乙醇混合后进行沉淀后冷冻干燥即得茯苓水溶性粗多糖,方法二为将所述样品通过微孔滤膜但不通过超滤膜部分,干燥即得茯苓水溶性粗多糖;更佳地,所述乙醇的体积占混合后溶液总体积的80%~90%。较佳地,所述酸水解为将酸性溶液和所得粗多糖混合后进行加热的步骤,所述加热的方式优选为水浴,所述加热的温度优选为90~100℃,更优选为90℃;所述加热的时间优选为4~6h,更优选为6h;所述酸性溶液优选为盐酸、三氟乙酸或硫酸。较佳地,所述衍生化处理包括与氢氧化钠溶液和1-苯基-3-甲基-5-吡唑啉酮的甲醇溶液混合后,再进行加热的步骤,所述1-苯基-3-甲基-5-吡唑啉酮的甲醇溶液与氢氧化钠溶液的体积比优选为大于等于1,所述加热的方式优选为水浴,所述加热的温度优选为60~70℃,更优选为70℃;所述加热的时间优选为30~40min,更优选为30min。较佳地,所述萃取的萃取剂为氯仿。为解决上述技术问题,本发明旨在提供一种茯苓质量的检测方法,所述检测方法为根据如权利要求1所述的构建方法生成待测茯苓样品的指纹图谱,所得指纹图谱具有甘露糖、核糖、葡萄糖、半乳糖、岩藻糖的5个特征峰,所述待测茯苓样品即为合格产品。较佳地,以所述葡萄糖的特征峰为对照,所述甘露糖、核糖、葡萄糖、半乳糖、岩藻糖5个特征峰的相对保留时间分别为0.50±5%min、0.61±5%min、1.00±5%min、1.16±5%min、1.47±5%min。较佳地,所述待测茯苓样品为茯苓汤剂或茯苓配方颗粒。为解决上述技术问题,本发明旨在提供一种检测茯苓多糖的单糖组成的方法,所述方法为利用高效液相色谱检测茯苓的供试品溶液;所述高效液相色谱的色谱柱为反相柱;所述高效液相色谱为使用流动相的体积比为15:85至20:80的乙腈-磷酸盐缓冲溶液进行等度洗脱;所述供试品溶液的制备包括:通过水煎煮法制得茯苓水溶性粗多糖;将所得粗多糖经酸水解后,以1-苯基-3-甲基-5-吡唑啉酮进行衍生化处理,萃取后取水层定容作为供试品溶液。较佳地,所述反相柱为硅胶键合相色谱柱,优选十八烷基硅烷键合色谱柱或辛烷基硅烷键合硅胶色谱柱硅胶柱。较佳地,所述磷酸盐缓冲溶液的ph值为6.5~7.0,优选6.80。较佳地,所述磷酸盐缓冲溶液的浓度为30~50mmol/l,优选50mmol/l。较佳地,所述磷酸盐缓冲溶液为kh2po4-naoh缓冲溶液。较佳地,所述流动相的流速为0.8~1.2ml/min,优选1ml/min。较佳地,所述反相柱的柱温为25~30℃,优选30℃。较佳地,所述高效液相色谱的检测波长为240~250nm,优选250nm。较佳地,所述流动相的体积比为18:82或17.5:82.5。较佳地,所述高效液相色谱的进样体积为10~20μl,优选10μl。较佳地,所述十八烷基硅烷键合色谱柱的柱长为150~250mm,优选250mm;和/或,所述十八烷基硅烷键合色谱柱的内径为2.1~4.6mm,优选4.6mm;和/或,所述十八烷基硅烷键合色谱柱的填料粒径为3~5μm,优选5μm;更佳地,所述十八烷基硅烷键合色谱柱为agilentextendc18,4.6*250mm,5μm。较佳地,所述水煎煮法包括以下步骤:(1)将茯苓通过水煎煮制得样品,(2)将(1)中所得样品进行后处理;所述后处理通过以下方法之一进行:方法一为将所述样品经浓缩或水溶,和乙醇混合后进行沉淀后冷冻干燥即得茯苓水溶性粗多糖,方法二为将所述样品通过微孔滤膜但不通过超滤膜部分,干燥即得茯苓水溶性粗多糖;更佳地,所述乙醇的体积占混合后溶液总体积的80%~90%。较佳地,所述酸水解为将酸性溶液和所得粗多糖混合后进行加热的步骤,所述加热的方式优选为水浴,所述加热的温度优选为90~100℃,更优选为90℃;所述加热的时间优选为4~6h,更优选为6h;所述酸性溶液优选为盐酸、三氟乙酸或硫酸。较佳地,所述衍生化处理包括与氢氧化钠溶液和1-苯基-3-甲基-5-吡唑啉酮的甲醇溶液混合后,再进行加热的步骤,所述1-苯基-3-甲基-5-吡唑啉酮的甲醇溶液与氢氧化钠溶液的体积比优选为大于等于1,所述加热的方式优选为水浴,所述加热的温度优选为60~70℃,更优选为70℃;所述加热的时间优选为30~40min,更优选为30min。较佳地,所述萃取的萃取剂为氯仿。为解决上述技术问题,本发明旨在提供一种茯苓多糖提取物的分离方法,其特征在于,所述方法为将通过水煎煮法获得的茯苓水溶性粗多糖经酸水解后,进行柱层析分离即可;所述柱层析为使用体积比为15:85至20:80的乙腈-磷酸盐缓冲溶液进行等度洗脱。较佳地,所述柱层析使用的层析柱为反相柱。较佳地,所述磷酸盐缓冲溶液的ph值为6.5~7.0,优选6.80。较佳地,所述磷酸盐缓冲溶液的浓度为30~50mmol/l,优选50mmol/l。较佳地,所述磷酸盐缓冲溶液为kh2po4-naoh缓冲溶液。较佳地,所述流动相的体积比为18:82或17.5:82.5。较佳地,所述水煎煮法包括以下步骤:(1)将茯苓通过水煎煮制得样品,(2)将(1)中所得样品进行后处理;所述后处理通过以下方法之一进行:方法一为将所述样品经浓缩或水溶,和乙醇混合后进行沉淀后冷冻干燥即得茯苓水溶性粗多糖,方法二为将所述样品通过微孔滤膜但不通过超滤膜部分,干燥即得茯苓水溶性粗多糖;更佳地,所述乙醇的体积占混合后溶液总体积的80%~90%。本发明中所述的茯苓为本领域常规的中药药材茯苓,例如购自中国各地的道地药材。本发明中所述1-苯基-3-甲基-5-吡唑啉酮(pmp)的甲醇溶液为本领域常规,主要是将1-苯基-3-甲基-5-吡唑啉酮(pmp)溶解在甲醇溶液中。其浓度为本领域常规,例如为0.5mol/l。在符合本领域常识的基础上,上述各优选条件,可任意组合,即得本发明各较佳实例。本发明所用试剂和原料均市售可得。本发明的积极进步效果在于:本发明所述茯苓质量的检测方法应用了能反映茯苓多糖的单糖组成特征的茯苓指纹图谱,可有效地检测分析茯苓汤剂及其配方颗粒的质量。本发明所述的茯苓指纹图谱的构建方法、茯苓质量的检测方法、检测茯苓多糖的单糖组成的方法以及茯苓多糖提取物的分离方法均操作简单、稳定、重现性好、检测效率高,具有较强实用性。附图说明图1a、图1b和图1c均为茯苓水溶性多糖的单糖组成hplc图谱;图1a为实施例4条件下的色谱图,图1b为实施例5条件下的色谱图,图1c为实施例6条件下的色谱图;图1a、图1b和图1c的色谱图中,峰1为甘露糖(man),峰2为核糖(rib),峰3为葡萄糖(glc),峰4为半乳糖(gal),峰5为岩藻糖(fuc)。图2为不同单糖参照物及其混合参照物的hplc色谱图,其中峰1为甘露糖(man),峰2为核糖(rib),峰3为葡萄糖(glc),峰4为半乳糖(gal),峰5为岩藻糖(fuc)。图3为不同产地22批次茯苓水溶性多糖的单糖组成hplc指纹图谱及系统生成的对照指纹图谱,图中s1至s22分别对应表1中药材批号bfl-001至bfl-022。图4为不同产地22批次茯苓构建的对照指纹图谱;其中峰1为甘露糖,峰2为核糖,峰3为葡萄糖,峰4为半乳糖,峰5为岩藻糖。图5为对比例1中茯苓碱溶性多糖的单糖组成hplc图谱,其中峰1为葡萄糖。图6为对比例2条件下的hplc色谱图;其中峰1为甘露糖,峰2为核糖,峰3为葡萄糖,峰4为半乳糖,峰5为岩藻糖。图7为对比例3条件下的hplc色谱图;其中峰1为甘露糖,峰2为核糖,峰3为葡萄糖,峰4为半乳糖,峰5为岩藻糖。具体实施方式下面通过实施例的方式进一步说明本发明,但并不因此将本发明限制在所述的实施例范围之中。下列实施例中未注明具体条件的实验方法,按照常规方法和条件,或按照商品说明书选择。实施例1:茯苓水溶性多糖的制备称取待测的茯苓饮片50g,加450ml水浸泡1小时,煮沸后再煎煮30min,过滤;药渣加350ml水煮沸后再煎煮25min,过滤,合并滤液。滤液减压浓缩至50ml,相当于每毫升有1g茯苓药材。50ml滤液中加入200ml无水乙醇(相当于80%醇沉),混匀,4℃冰箱静置过夜,离心,沉淀冷冻干燥,即得茯苓水溶性粗多糖。实施例2:茯苓水溶性多糖的单糖组成指纹图谱分析的供试品溶液的制备称取实施例1制得的茯苓水溶性粗多糖约20mg,加入5ml4mol/l的hcl溶液,90℃水浴6h;取出,加入适量甲醇进行减压浓缩,重复操作数次以除去盐酸,残渣加水定容至1ml,得水解液,备用;吸取所得水解液100μl,加入50μl0.3mol/l的naoh溶液,再加入60μl0.5mol/l1-苯基-3-甲基-5-吡唑啉酮(pmp)甲醇溶液,70℃水浴30min,取出,冷却至室温,加入50μl0.3mol/l的hcl溶液中和。加入适量氯仿萃取数次,取水层定容至1ml,作为供试品溶液。实施例3:单糖参照物的对照品溶液的制备不同单糖参照物的对照品溶液的制备:精密称取对照品甘露糖(d-man,批号f20120107,国药集团化学试剂有限公司)、核糖(d-rib,批号1451793,sigma)、葡萄糖(glc,批号20070115,国药集团化学试剂有限公司)、半乳糖(d-gal;批号840215;上海试剂二厂)、岩藻糖(d-fuc,050m1909,sigma)各10mg,分别加水溶解配制成1mg/ml的单糖贮备液。精密移取各单糖储备液100μl,加入50μl0.3mol/l的naoh溶液,再加入60μl0.5mol/l的pmp甲醇溶液,70℃水浴30min,取出,冷却至室温,加入50μl0.3mol/l的hcl溶液中和。加入适量氯仿萃取数次,取水层定容至1ml,制得不同单糖的衍生化对照品溶液(浓度约0.1mg/ml)。混合参照物溶液的制备:分别移取上述的各单糖储备液100μl,混合均匀得初步的混合参照物溶液,精密移取初步的混合参照物溶液100μl,加入50μl0.3mol/l的naoh溶液,再加入60μl0.5mol/l的pmp甲醇溶液,70℃水浴30min,取出,冷却至室温,加入50μl0.3mol/l的hcl溶液中和。加入适量氯仿萃取数次,取水层定容至1ml,制得混合参照物溶液(浓度约0.02mg/ml)。不同单糖参照物及其混合参照物的hplc色谱条件参照实施例5,即:extendc18色谱柱(5μm,4.6×250mm,agilent),以乙腈-50mmol/l磷酸盐缓冲液水(ph=6.80)(18:82,v/v)为流动相,其中磷酸盐缓冲溶液为kh2po4-naoh缓冲溶液,流速1.0ml/min,检测波长250nm,柱温:30℃;进样体积10μl。所得结果如图2所示。实施例4:茯苓水溶性多糖的单糖组成指纹图谱分析的液相色谱系统1取extendc18色谱柱(5μm,4.6×250mm,agilent),以乙腈-50mmol/l(混合前磷酸缓冲液的浓度)磷酸盐缓冲液水(ph=6.80)(20:80,v/v)为流动相,其中磷酸盐缓冲溶液为kh2po4-naoh缓冲溶液,流速1.0ml/min,检测波长250nm,柱温:30℃;进样体积10μl。实施例5:茯苓水溶性多糖的单糖组成指纹图谱分析的液相色谱系统2取extendc18色谱柱(5μm,4.6×250mm,agilent),以乙腈-50mmol/l磷酸盐缓冲液水(ph=6.80)(18:82,v/v)为流动相,其中磷酸盐缓冲溶液为kh2po4-naoh缓冲溶液,流速1.0ml/min,检测波长250nm,柱温:30℃;进样体积10μl。实施例6:茯苓水溶性多糖的单糖组成指纹图谱分析的液相色谱系统3取extendc18色谱柱(5μm,4.6×250mm,agilent),以乙腈-50mmol/l磷酸盐缓冲液水(ph=6.80)(17.5:82.5,v/v)为流动相,其中磷酸盐缓冲溶液为kh2po4-naoh缓冲溶液,流速1.0ml/min,检测波长250nm,柱温:30℃;进样体积10μl。按照实施例1和2所述的制备方法制备茯苓水溶性多糖的单糖组成指纹图谱分析的供试品溶液,茯苓购于安徽石田中药饮片有限公司,货号1802005。所得hplc的结果如图1所示。其中a为实施例4条件下的色谱图,b为实施例5条件下的色谱图,c为实施例6条件下的色谱图;与图2的单糖参照物比对得知,其中峰1为甘露糖,峰2为核糖,峰3为葡萄糖,峰4为半乳糖,峰5为岩藻糖。图中显示,3种条件下的5个特征峰都能得到很好的分离,并且实施例6的条件下所得图谱分离度最好。实施例7:22批茯苓水溶性多糖的单糖组成指纹图谱的建立及共有特征峰的指认取茯苓样品22批次,按照实施例1和2所述的制备方法制备茯苓水溶性多糖的单糖组成指纹图谱分析的供试品溶液,按实施例6条件进行检测,所得的hplc指纹图谱结果如图3所述。将得到的22批茯苓样品的色谱图以aia格式导入中药色谱指纹图谱相似度评价系统(2004a版)进行相似度评价,确定其有5个特征共有峰,与图2的单糖参照物比对得知,这5个共有特征峰依次为:甘露糖、核糖、葡萄糖、半乳糖、岩藻糖。22批茯苓样品的图谱的保留时间、以葡萄糖(即图4中的3号峰)为对照的相对保留时间、峰面积平均值汇总如下(相应的对照指纹图谱如图4所示):峰1为甘露糖,平均保留时间rt为10.286min,相对保留时间为0.50min,平均峰面积为26.082mau,rsd值为44.60%;峰2为核糖,平均保留时间rt为12.519min,相对保留时间为0.61min,平均峰面积为2.250mau,rsd值为30.44%;峰3为葡萄糖,平均保留时间rt为20.606min,相对保留时间为1.00min,平均峰面积为92.315mau,rsd值为50.64%;峰4为半乳糖,平均保留时间rt为23.82min,相对保留时间为1.16min,平均峰面积为41.551mau,rsd值为53.84%;峰5为岩藻糖,平均保留时间rt为30.293min,相对保留时间为1.47min,平均峰面积为4.630mau,rsd值为61.34%。保留时间rt为9min处为残留的衍生化试剂pmp,5min之前为溶剂峰。22批茯苓样品的单糖指纹图谱的相似度分析结果如表1所示,除样品bfl-013的相似度为0.89外,其他21批次的样品的相似度均大于0.9。从而说明了本发明所述分析方法能够较好的表征茯苓的特征,对茯苓质量评价有较好的参考价值。表1不同产地22批次茯苓水溶性多糖的单糖组成相似度分析结果药材批号商购来源货号产地相似度bfl-001上海雷允上药业西区有限公司170508安徽0.929bfl-002上海雷允上药业西区有限公司170602安徽0.947bfl-003金牛区(成都)荷花池中药材专业市场芯草中药行茯苓3-1云南0.976bfl-004金牛区(成都)荷花池中药材专业市场芯草中药行茯苓3-2云南0.972bfl-005金牛区(成都)荷花池中药材专业市场芯草中药行茯苓3-3云南0.91bfl-006沈阳维康医药连锁有限公司170201安徽0.999bfl-007太安堂中药饮片有限公司170803安徽亳州0.955bfl-008太安堂中药饮片有限公司170724安徽大别山0.981bfl-009太安堂中药饮片有限公司170807云南0.991bfl-010太安堂中药饮片有限公司170806湖北0.996bfl-011由神威药业提供1708312安徽岳西0.999bfl-012由神威药业提供1708311安徽岳西0.994bfl-013由神威药业提供1708314安徽岳西0.89bfl-014由神威药业提供1708315安徽岳西0.948bfl-015神威药业提供1708313安徽岳西0.981bfl-016安徽石田中药饮片有限公司1802003安徽岳西0.986bfl-017安徽石田中药饮片有限公司1802002陕西0.974bfl-018安徽石田中药饮片有限公司1802006安徽0.947bfl-019安徽石田中药饮片有限公司1802001云南0.962bfl-020安徽石田中药饮片有限公司1802007安徽大别山0.999bfl-021安徽石田中药饮片有限公司1802004安徽金寨0.994bfl-022安徽石田中药饮片有限公司1802005云南0.977对比例对比例1:茯苓碱溶性粗多糖的提取取茯苓粉末(购于安徽石田中药饮片有限公司,货号1802005)1g,溶于3℃的0.5mol/l氢氧化钠溶液中,搅拌至粉末溶解。5℃以下冷藏过夜,然后离心,所得滤液以10%(v/v)醋酸液中和至酸性,再加入等体积的95%(v/v)乙醇,于3℃放置过夜,然后离心,沉淀冷冻干燥,即得茯苓碱溶性粗多糖。将所得碱溶性粗多糖按照实施例2所述制备方法制备指纹图谱分析的供试品溶液,按照实施例5的hplc条件进行检测,所得多糖的单糖组成的hplc结果如图5所示,结果表明,按该对比例的碱提法得到的多糖为均多糖,单糖组成仅为葡萄糖一种糖,不能表征茯苓的特征性,而本发明所用的水溶性提取得到的多糖由5种单糖组成,能够较好的表征茯苓的特征性。对比例2:茯苓水溶性多糖的单糖组成指纹图谱分析的液相色谱系统4取extendc18色谱柱(5μm,4.6×250mm,agilent),流速1.0ml/min,检测波长250nm,柱温30℃;进样体积10μl;流动相:溶剂a,15%(乙腈体积占两者混合后总体积的体积百分比)乙腈+50mmol/l(两者混合前磷酸缓冲液的浓度)磷酸缓冲液(ph=6.8);溶剂b,40%乙腈+50mmol/l磷酸缓冲液(ph6.8)。梯度为0~10min:17%b;10~20min:17~20%b;20~25min:20%b;25~30min:20~21%b;30~35min:21~22%b;35~35.1min:22~17%b;35.1~45min:17%b。按照实施例1和2所述方法制备茯苓水溶性多糖的单糖组成指纹图谱分析的供试品溶液,茯苓购于安徽石田中药饮片有限公司;货号1802005,按照该实施例所述的hplc条件进行检测,所得色谱图如图6所示。结果显示,3号峰(葡萄糖)和4号峰(半乳糖)不能完全分离,且流动相的配制较繁琐。而利用本发明所述条件能将3号峰和4号峰完全分离,有良好的分离度,且流动相配置相对于此实施例较简单。对比例3:茯苓水溶性多糖的单糖组成指纹图谱分析的液相色谱系统5取extendc18色谱柱(5μm,4.6×250mm,agilent),流速1.0ml/min,检测波长250nm,柱温30℃;进样体积10μl;流动相:乙腈(b)-0.1%磷酸(a);洗脱梯度:0~10min:15%~15%b;10~50min:15~30%b。按照实施例1和实施例3制备得到混合对照品溶液,按照该实施例所述的hplc条件进行检测,所得色谱图如图7所示。图中显示:2号峰和3号峰、4号峰和5号峰均不能实现较好的分离。当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1