一种鉴别洋槐蜜真实性的方法与流程
本发明属于食品检验技术领域,尤其涉及一种鉴别洋槐蜜真实性的方法。
背景技术:
洋槐蜜作为我国产量最大的单花蜜之一,深受消费者的喜爱。然而,市场上伪劣掺假的洋槐蜜蜂蜜非常普遍,洋槐蜜蜂蜜造假已经形成规模化、专业化。大量假洋槐蜜混入市场,严重威胁消费者的健康,影响我国蜂产业的发展。
目前鉴别蜂蜜真实性的方法主要有;包括感官鉴别、花粉分析以及理化指标测试。近年来,利用先进的现代仪器设备,针对蜂蜜掺假发展了多种检测分析技术和方法,主要包括稳定碳同位素比率法(gb/t18932.1–2002)、色谱法(gb/t18932.1–2002)光谱分析(近红外、荧光、拉曼、紫外-可见吸收光谱)、淀粉酶检测法等。这些方法存在结果不稳定、操作复杂,成本高等问题。
技术实现要素:
有鉴于此,本发明提供了一种鉴别洋槐蜜真实性的方法,本发明所述的方法,结果可靠稳定、判断度高、方便操作、简单易行、成本低廉。
基于上述目的,本发明提供了一种鉴别洋槐蜜真实性的方法,包括以下步骤:1)将待鉴蜂蜜与水混合溶解,调节ph值为6~7,获得样品溶液;2)将所述样品溶液过反相-阴离子交换固相萃取柱,用含10wt%甲酸的甲醇溶液洗脱萃取柱,收集洗脱液;3)用高效液相色谱检测所述洗脱液,将检测到的物质图谱与洋槐蜜多酚类成分的指纹图谱比对,若检测到物质的种类与洋槐蜜多酚类成分的指纹图谱一致则为真洋槐蜜;所述洋槐蜜多酚类成分的指纹图谱为包括红花菜豆酸、反,反式脱落酸和顺,反式脱落酸的指纹图谱。
优选的,所述步骤3)还包括高效液相色谱分离后,根据高效液相色谱分离结果用外标法计算检测到物质的含量。
优选的,所述红花菜豆酸在真洋槐蜜中的含量>25μg/100g。
优选的,所述反,反式脱落酸在真洋槐蜜中的含量>15μg/100g。
优选的,所述顺,反式脱落酸在真洋槐蜜中的含量>50μg/100g。
优选的,步骤3)中所述的高效液相色谱的流速为0.7ml/min;紫外检测波长为280nm;分离柱为c18柱,所述c18柱的规格为150×4.6mm,5μm;上样体积为20μl;
所述的高效液相色谱的流动相a为含质量分数0.2%乙酸的水;流动相b为含质量分数0.2%乙酸的甲醇;
洗脱梯度程序为:[0~11)min,[9%~14%)流动相b;[11~14)min,[14%~15%)流动相b;[14~17)min,15%流动相b;[17~24)min,(15%~16%]流动相b;[24~28)min,(16%~17%]流动相b;[28~30)min,(17%~22%]流动相b;[30~38)min,(22%~25%]流动相b;[38~41)min,(25%~30%]流动相b;[41~46)min,(30%~33%]流动相b;[46~55)min,33%流动相b;[55~60)min,(33%~34%]流动相b;[60~70)min,(34%~36%]流动相b;[70~80)min,(36%~40%]流动相b;[80~90)min,(40%~45%]流动相b;[90~100)min,(45~52%]流动相b。
优选的,所述洋槐蜜多酚类物质指纹图谱通过以下方法获得:取100~150份真洋槐蜜样品,分别进行高效液相色谱分析,得到每一份样品的多酚类成分色谱图,将所有样品的多酚类成分色谱图导入中药指纹图谱软件,获得洋槐蜜中多酚类成分的指纹图谱。
优选的,步骤3)中外标法的标准曲线为y=4513x+135049,其中x为检测物质的峰面积,y检测物质的含量。
优选的,所述待鉴样品与水的质量比为1:(3.5~4.5)。
优选的,步骤2)获得洗脱液后,在进行高效液相色谱检测前,将所述洗脱液过滤。
本发明的有益效果:本发明提供的鉴别洋槐蜜真实性的方法,将待鉴样品与水混合溶解后过反相-阴离子交换固相萃取柱,对待鉴样品中的多酚类物质进行富集,洗脱液中的多酚类物质更容易被检测到,检测结果可靠稳定、判断度高、方便操作、成本低廉。
附图说明
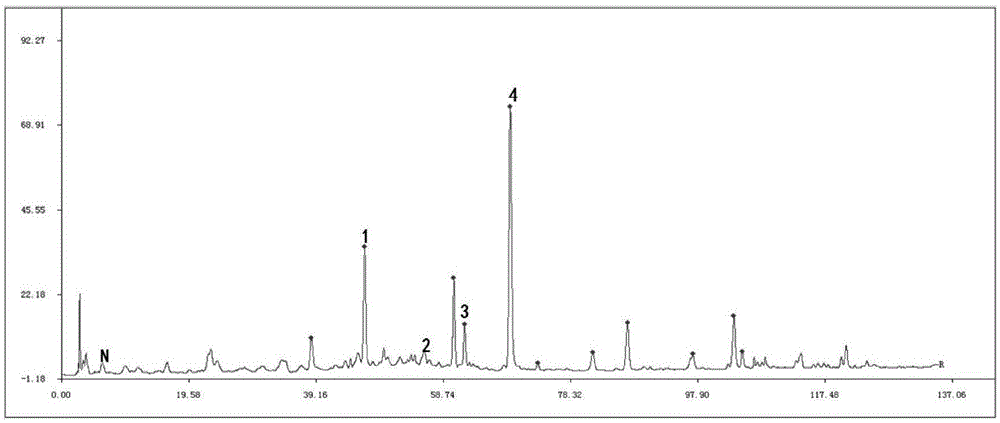
图1为洋槐蜜多酚类成分的指纹图谱。
具体实施方式
本发明提供了一种鉴别洋槐蜜真实性的方法,包括以下步骤:1)将待鉴蜂蜜与水混合溶解,调节ph值为6~7,获得样品溶液;2)将所述样品溶液过反相-阴离子交换固相萃取柱,用含10wt%甲酸的甲醇溶液洗脱萃取柱,收集洗脱液;3)用高效液相色谱检测所述洗脱液,将检测到的物质图谱与洋槐蜜多酚类成分的指纹图谱比对,若检测到物质的种类与洋槐蜜多酚类成分的指纹图谱一致则为真洋槐蜜;所述洋槐蜜多酚类成分的指纹图谱为包括红花菜豆酸、反,反式脱落酸和顺,反式脱落酸的指纹图谱。
在本发明中,将待鉴样品与水混合溶解,调节ph值为6~7,获得样品溶液。在本发明中,所述待鉴样品与水的质量比优选为1:(3.5~4.5),更优选为1:(3.8~4.2),最优选的为1:4;所述在本发明中,所述待鉴样品为任何形式的蜂蜜,包括但不限于原料蜜和商品蜜;所述水优选为超纯水。在本发明中优选的采用碱类物质调节溶液的ph值,更优选的采用氨水调节溶液的ph值;所述氨水的质量浓度优选为4~6%,更优选为5%。
本发明在获得样品溶液后,将所述样品溶液过反相-阴离子交换固相萃取柱,收集获得洗脱液。本发明中所述样品溶液在过反相-阴离子交换固相萃取柱之前,优选的进行固液分离;所述固液分离的方式优选为离心,所述离心的离心力优选为9000~11000×g,更优选为10000×g,所述离心的时间优选为8~12min,更优选为10min。本发明中,所述反相-阴离子交换固相萃取柱的规格型号strata-x-a。本发明所述的反相-阴离子交换固相萃取柱在使用前优选的经甲醇活化后用超纯水平衡。在本发明中所述甲醇的用量优选为0.8~1.2ml,更优选为1.0ml;所述超纯水的用量优选为0.8~1.2ml,更优选为1.0ml。在本发明中,将所述样品溶液上柱后,优选的用超纯水清洗萃取柱,然后用含10wt%甲酸的甲醇溶液洗脱萃取柱,收集洗脱液。在本发明中,所述清洗萃取柱用的超纯水的用量优选为0.8~1.2ml,更优选为1.0ml;所述含10wt%甲酸的甲醇溶液的用量为1.5~2.5ml,更优选为2.0ml。本发明在获得洗脱液后,优选的还包括将所述洗脱液干燥后复溶。在本发明中,所述干燥优选的采用氮气吹干;本发明对所述氮气的流速和氮气吹干的时间没有特殊限定,能够实现干燥即可。本发明在所述干燥后,优选的采用色谱级乙酸甲醇溶液(2:98v/v)进行复溶,所述色谱级乙酸甲醇溶液的用量优选为1.5~2.5ml,更优选为2ml。本发明在获得复溶的洗脱液后,在进行高效液相色谱检测前,将所述洗脱液过滤。所述过滤优选为滤膜过滤,所述滤膜优选为0.22μm滤膜,过滤后得到高效液相色谱上样液。
本发明在获得高效液相色谱上样液后,用高效液相色谱检测所述上样液液,将检测到的物质与洋槐蜜多酚类成分的指纹图谱比对,检测到物质的种类与洋槐蜜多酚类成分的指纹图谱一致则为真洋槐蜜。
在本发明中所述高效液相色谱的流动相a为含质量分数0.2%乙酸的水;流动相b为含质量分数0.2%乙酸的甲醇;所述的高效液相色谱的流速为0.7ml/min;紫外检测波长为280nm;分离柱为c18柱,所述c18柱的规格为150×4.6mm,5μm;上样体积为20μl;洗脱梯度为[0~11)min,[9%~14%)流动相b;[11~14)min,[14%~15%)流动相b;[14~17)min,15%流动相b;[17~24)min,(15%~16%]流动相b;[24~28)min,(16%~17%]流动相b;[28~30)min,(17%~22%]流动相b;[30~38)min,(22%~25%]流动相b;[38~41)min,(25%~30%]流动相b;[41~46)min,(30%~33%]流动相b;[46~55)min,33%流动相b;[55~60)min,(33%~34%]流动相b;[60~70)min,(34%~36%]流动相b;[70~80)min,(36%~40%]流动相b;[80~90)min,(40%~45%]流动相b;[90~100)min,(45~52%]流动相b。本发明在所述高效液相色谱检测后,将所述高效液相色谱检测到的物与洋槐蜜多酚类成分的指纹图谱比对,确定检测到物质的种类。在本发明中,所述洋槐蜜多酚类物质指纹图谱通过以下方法获得:取100~150份真洋槐蜜样品,进行高效液相色谱,得到每一份样品的多酚类成分色谱图,将所有样品的多酚类成分色谱图导入中药指纹图谱软件,获得洋槐蜜中多酚类成分的指纹图谱。本发明中,所述洋槐蜜多酚类成分的指纹图谱优选的如图1所示,其中1为红花菜豆酸,3为反,反式脱落酸,4为顺,反式脱落酸。
本发明在确定检测到物质的种类后,使用外标法计算含量;所述反,反式脱落酸和顺,反式脱落酸的标准曲线为y=4513x+135049,其中x为检测物质的峰面积,y检测物质的含量;本发明中,y的单位为纳克(ng)。本发明中所述红花菜豆酸以脱落酸的标准曲线来计算,由于红花菜豆酸无标准物质,且所述红花菜豆酸是脱落酸的衍生物,结构相似,因此用脱落酸计。本发明中,所述红花菜豆酸在真洋槐蜜中的含量优选的>25μg/100g;所述反,反式脱落酸在真洋槐蜜中的含量优选的>15μg/100g;所述顺,反式脱落酸在真洋槐蜜中的含量优选的>50μg/100g。
下面结合实施例对本发明提供的技术方案进行详细的说明,但是不能把它们理解为对本发明保护范围的限定。
实施例1
某中间商对收集的原料洋槐蜜蜜进行质量评估。
操作方法:
①准确称取20g洋槐蜜蜂蜜样品,置于干燥的瓶中,加入80ml超纯水溶解;
②用5%的氨水调节ph至6—7;
③10000×g离心10min,取上清备用;
④反相-阴离子交换固相萃取柱(rp-aespe)经1ml甲醇活化后用1ml超纯水(ph:6-7)平衡,上清液过柱;
⑤1ml超纯水清洗萃取柱,2ml甲醇(含10%甲酸)洗脱萃取柱,收集洗脱液;
⑥洗脱液在室温条件下用氮气吹至干燥,用2ml色谱级乙酸甲醇溶液(2:98v/v)复溶混匀,过0.22μm滤膜,得到hplc上样液。
⑦使用hplc方法检测所述hplc上样液的;高效液相色谱法(hplc)的检测方法为:流动相a为水(含0.2%乙酸);流动相b为甲醇(含0.2%乙酸);总流速为0.7ml/min;紫外检测波长为280nm;分离柱为c18柱,(150×4.6mm,5μm);上样体积为20μl;洗脱梯度为0-11min,9%-14%b;11-14min,14-15%b;14-17min,15%b;17-24min,15-16%b;24-28min,16-17%b;28-30min,17-22%b;30-38min,22-25%b;38-41min,25-30%b;41-46min,30-33%b;46-55min,33%b;55-60min,33-34%b;60-70min,34-36%b;70-80min,36-40%b;80-90min,40-45%b;90-100min,45-52%b。
⑧使用外标法计算含量:
反,反式脱落酸和顺,反式脱落酸的标准曲线为y=4513x+135049;红花菜豆酸以脱落酸计。
按照hplc方法对上样液进行定性定量分析:
按照标曲进行定量分析,得到红花菜豆酸为60μg/100g;反,反式脱落酸为40μg/100g;顺,反式脱落酸为90μg/100g。并且色谱图与标准指纹图谱相比,无其他异常物质。综上,该原料蜜是洋槐蜜。
实施例2
某企业对采购的原料蜜进行质量评估。
操作方法:
①准确称取20g洋槐蜜蜂蜜样品,置于干燥的瓶中,加入80ml超纯水溶解;
②用5%的氨水调节ph至6—7;
③10000×g离心10min,取上清备用;
④反相-阴离子交换固相萃取柱(rp-aespe)经1ml甲醇活化后用1ml超纯水(ph:6-7)平衡,上清液过柱;
⑤1ml超纯水清洗萃取柱,2ml甲醇(含10%甲酸)洗脱萃取柱,收集洗脱液;
⑥洗脱液在室温条件下用氮气吹至干燥,用2ml色谱级乙酸甲醇溶液(2:98v/v)复溶混匀,过0.22μm滤膜,得到hplc上样液。
⑦使用hplc方法检测所述hplc上样液的;高效液相色谱法(hplc)的检测方法为:流动相a为水(含0.2%乙酸);流动相b为甲醇(含0.2%乙酸);总流速为0.7ml/min;紫外检测波长为280nm;分离柱为c18柱,(150×4.6mm,5μm);上样体积为20μl;洗脱梯度为0-11min,9%-14%b;11-14min,14-15%b;14-17min,15%b;17-24min,15-16%b;24-28min,16-17%b;28-30min,17-22%b;30-38min,22-25%b;38-41min,25-30%b;41-46min,30-33%b;46-55min,33%b;55-60min,33-34%b;60-70min,34-36%b;70-80min,36-40%b;80-90min,40-45%b;90-100min,45-52%b。
⑧使用外标法计算含量:
反,反式脱落酸和顺,反式脱落酸的标准曲线为y=4513x+135049;红花菜豆酸因没有标准物质,因此红花菜豆酸的标曲以脱落酸的记。
按照hplc方法对上样液进行定性定量分析:
按照标曲进行定量分析,得到红花菜豆酸为50μg/100g;反,反式脱落酸为42μg/100g;顺,反式脱落酸为79μg/100g。并且色谱图与标准指纹图谱相比,无其他异常物质。综上,该原料蜜是洋槐蜜。
实施例3
某第三方检测机构对委托检测的商品蜜进行质量检测。
操作方法:
操作方法:
①准确称取20g洋槐蜜蜂蜜样品,置于干燥的瓶中,加入80ml超纯水溶解;
②用5%的氨水调节ph至6—7;
③10000×g离心10min,取上清备用;
④反相-阴离子交换固相萃取柱(rp-aespe)经1ml甲醇活化后用1ml超纯水(ph:6-7)平衡,上清液过柱;
⑤1ml超纯水清洗萃取柱,2ml甲醇(含10%甲酸)洗脱萃取柱,收集洗脱液;
⑥洗脱液在室温条件下用氮气吹至干燥,用2ml色谱级乙酸甲醇溶液(2:98v/v)复溶混匀,过0.22μm滤膜,得到hplc上样液。
⑦使用hplc方法检测所述hplc上样液的;高效液相色谱法(hplc)的检测方法为:流动相a为水(含0.2%乙酸);流动相b为甲醇(含0.2%乙酸);总流速为0.7ml/min;紫外检测波长为280nm;分离柱为c18柱,(150×4.6mm,5μm);上样体积为20μl;洗脱梯度为0-11min,9%-14%b;11-14min,14-15%b;14-17min,15%b;17-24min,15-16%b;24-28min,16-17%b;28-30min,17-22%b;30-38min,22-25%b;38-41min,25-30%b;41-46min,30-33%b;46-55min,33%b;55-60min,33-34%b;60-70min,34-36%b;70-80min,36-40%b;80-90min,40-45%b;90-100min,45-52%b。
⑧使用外标法计算含量:
反,反式脱落酸和顺,反式脱落酸的标准曲线为y=4513x+135049;红花菜豆酸因没有标准物质,因此红花菜豆酸的标曲以脱落酸的记。
按照hplc方法对上样液进行定性定量分析:
按照标曲进行定量分析,得到红花菜豆酸为55μg/100g;反,反式脱落酸为32μg/100g;顺,反式脱落酸为69μg/100g。并且色谱图与标准指纹图谱相比,无其他异常物质。综上,该原料蜜是洋槐蜜。
由上述实施例可知,本发明提供的鉴别洋槐蜜真实性的方法,检测结果可靠稳定、判断度高、方便操作、成本低廉。
以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!