腰肾膏指纹图谱的建立方法及腰肾膏对照指纹图谱与流程
本发明涉及药物分析领域,主要涉及一种腰肾膏指纹图谱的建立方法及腰肾膏对照指纹图谱。
背景技术:
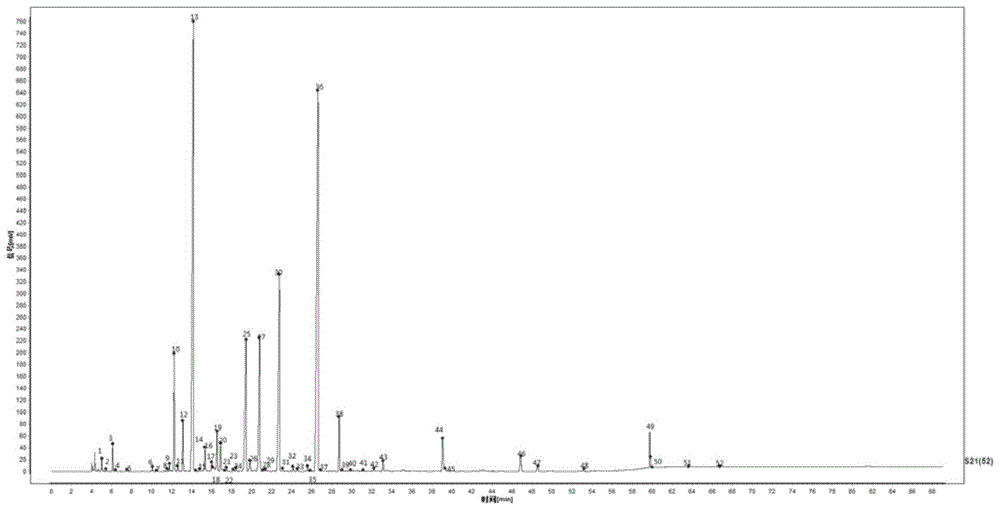
:腰肾膏处方含肉苁蓉、八角茴香、熟地黄、补骨脂、淫羊藿、蛇床子、牛膝、续断、甘草、杜仲等二十种药材,加上樟脑、冰片、薄荷素油等,辅料为白凡士林、橡胶、萜烯树脂等。本品具有温肾助阳,强筋壮骨,祛风止痛的作用。用于肾虚性腰膝酸痛,肌肉酸痛,夜尿。由于其成分多而复杂,目前缺乏全面反映其质量和真伪的方法,且目前并未有关于腰肾膏指纹图谱的研究报道,因而,构建针对腰肾膏指纹图谱的研究,对产品质量控制具有较大的意义,能够为腰肾膏提供一种全面的质量控制方法。因此,现有技术还有待于改进和发展。技术实现要素:鉴于上述现有技术的不足,本发明的目的在于提供一种腰肾膏指纹图谱的建立方法及腰肾膏对照指纹图谱,通过对腰肾膏指纹图谱的研究,得到了一种较好的腰肾膏指纹图谱建立方法以及标准指纹图谱,为控制腰肾膏质量提供了新的技术方法,旨在解决目前对腰肾膏的质量控制技术不足的问题。本发明的技术方案如下:经过了对样品的前处理和其测定条件的摸索,本发明方案中将成分分为挥发性成分和不挥发性成分两种类别,其中挥发油成分通过乙酸乙酯进行收集,用乙酸乙酯收集液进行气相分析;剩余膏体用50%甲醇进行超声提取,浸液进行液相分析。通过使用对照品、阴性对照和对照药材与指纹图谱进行对比,对指纹图谱中主要共有峰进行了归属,由此建立腰肾膏的两个指纹图谱。得到的指纹图谱使用中药指纹图谱相似度评价系统进行分析,进行客观评价。结合液相和气相指纹图谱能比较全面的反映腰肾膏中有效成分的组成,有助于腰肾膏的质量控制。具体地,所述腰肾膏指纹图谱的建立方法,包括以下步骤:(1)样品的前处理:用乙酸乙酯吸收腰肾膏内的挥发性成分,作为气相鉴别用供试品;剩余膏体加水,经50%甲醇超声提取,浸液作为液相鉴别供试品。具体地,所述气相鉴别用供试品的提取过程为:将腰肾膏塞入烧瓶中,加水,接上载有乙酸乙酯的挥发油提取器,在电热套上回流;回流完毕后放冷,将挥发油提取器的乙酸乙酯层放入分液漏斗中,用乙酸乙酯冲洗挥发油提取器,洗液并入分液漏斗中,弃去水层,乙酸乙酯层通过载有无水硫酸钠的滤纸,收集滤液,加乙酸乙酯至刻度,摇匀,得到气相鉴别用供试品。更具体地,可以为:取三片腰肾膏剪成条状,撕去背衬,以一片为单位结合成团,塞入250ml烧瓶中,加水100ml,接上载有2ml乙酸乙酯的挥发油提取器,在电热套上回流3小时;回流完毕后放冷,将挥发油提取器的乙酸乙酯层放入分液漏斗中,分别用乙酸乙酯2ml、2ml、1ml冲洗挥发油提取器,洗液并入分液漏斗中,弃去水层,乙酸乙酯层通过载有1g无水硫酸钠的滤纸,滤入10ml容量瓶中,加乙酸乙酯至刻度,摇匀,得到气相鉴别用供试品。所述液相鉴别用供试品的提取过程为:所述液相鉴别用供试品的提取过程为:提取后的膏体弃去水浸液,用纯化水荡洗,烘干,取出膏体剪碎,置于具塞容量瓶中,加入50%甲醇,超声处理,放冷,浸液用0.45μm微孔滤膜过滤,滤液作为液相鉴别用供试品。更具体地,可以为:提取后的膏体弃去水浸液,用100ml纯化水荡洗,重复三次,在105℃烘干,取其中一片的量,剪碎,置于50ml具塞锥形瓶中,加入50%甲醇30ml,超声60min,放冷,浸液用0.45μm微孔滤膜过滤,滤液作为液相鉴别用供试品。(2)对气相鉴别用供试品进行气相分析,得到腰肾膏气相指纹图谱;色谱柱采用zb-wax气相色谱柱;进样口温度为200℃;检测器温度为280℃;流速为1ml/min;分流比为10:1;进样量为1μl;升温速率设置为,初始温度为90℃,分析过程中,保持7分钟,随后以每分钟10℃的速率升温至115℃,保持10分钟,随后以每分钟3℃的速率升温至150℃,保持0分钟,随后以每分钟0.5℃的速率升温至160℃,保持0分钟,随后以每分钟10℃的速率升温至240℃,保持30分钟。(3)对液相鉴别用供试品进行液相分析,得到腰肾膏液相指纹图谱;色谱柱采用welchultimatexb-c18液相色谱柱,内径4.6mm*柱长250mm,填料粒径为5μm;检测波长为245nm,进样量为20μl;流动相由有机相和水相组成,有机相为乙腈,水相为0.02%三氟乙酸溶液;洗脱条件为0~35min,有机相:水相=15:85;35~55min,有机相:水相=47:53;55~65min,有机相:水相=47:53;65~80min,有机相:水相=50:50;80~87min,有机相:水相=65:35;87~97min,有机相:水相=73:27;97~119min,有机相:水相=100:0;理论塔板数以蛇床子素峰计算,不少于10000。所述的腰肾膏指纹图谱的建立方法,其中,还包括以下步骤:(4)将得到的指纹图谱与对照指纹图谱进行相似度计算:将得到的气相指纹图谱与气相对照指纹图谱进行相似度计算;将得到的液相指纹图谱与液相对照指纹图谱进行相似度计算。按中药色谱指纹图谱相似度评价系统,可用于供试品质量的整体评价,供试品指纹图谱与对照图谱经相似度计算,相似度不得低于0.90,且应具有相应的药材特征峰。本发明中还提供腰肾膏对照指纹图谱,分为腰肾膏气相对照指纹图谱和腰肾膏液相对照指纹图谱;该腰肾膏的气相对照指纹图谱和液相对照指纹图谱的建立,可作为腰肾膏的整体质量控制方法。腰肾膏气相对照指纹图谱如图1所示,以水杨酸甲酯作为参照峰,得到了51个共有峰,对其中20个共有峰进行了药材归属:13号峰为樟脑,36号峰为水杨酸甲酯;1号峰、2号峰、3号峰、4号峰、10号峰、11号峰、12号峰、16号峰、17号峰、25号峰、26号峰为薄荷素油特征峰;27号峰、30号峰为冰片特征峰;44号峰为肉桂油特征峰;19号峰为枫香脂特征峰;46号峰、48号峰为丁香特征峰;38号峰为八角茴香和小茴香共有特征峰。腰肾膏液相对照指纹图谱如图2所示,以蛇床子素作为参照峰,得到了52个共有峰,对其中21个共有峰进行了药材归属:20号,24号,25号,30号,39号,40号峰为五味子特征峰;15号,21号,23号,27号,28号峰为蛇床子特征峰;13号,14号,22号,26号,31号,42号峰为补骨脂特征峰;12号,37号,38号峰为枫香脂特征峰;49号峰为乳香特征峰。有益效果:本发明中,利用中药色谱指纹图谱相似度评价系统建立腰肾膏的气相指纹图谱和液相指纹,指纹图谱相似度良好(≥0.90)。因此,本发明建立的腰肾膏质量标准稳定可靠,可用于腰肾膏的的质量控制。本发明腰肾膏的指纹图谱构建方法,具有以下显著的优点:(1)因腰肾膏成分复杂,本发明将腰肾膏成分分为挥发性成分和不挥发性成分两种类别,对腰肾膏分别进行指纹图谱研究,为腰肾膏质量控制提供了新的分析方法。(2)优化指纹图谱的构建方法,使得到的指纹图谱基线平稳、分离度好、峰型较好、出峰量多。(3)本发明供试样品制作简单,色谱条件容易实现,精密度高,而且其稳定性和重现性较好,本发明采用的检测波长出峰较多,反映的信息完全。附图说明图1为腰肾膏气相对照指纹图谱(带标号)。图2为腰肾膏液相对照指纹图谱(带标号)。图3为指认对照品特征峰的液相指纹图谱。图4为指认冰片对照品特征峰的气相指纹图谱。图5为指认肉桂油对照品特征峰的气相指纹图谱。图6为指认枫香脂对照品特征峰的气相指纹图谱。图7为指认丁香对照品特征峰的气相指纹图谱。图8为指认八角茴香和小茴香对照品特征峰的气相指纹图谱。图9为腰肾膏液相指纹图谱建立方法采用三根同一品牌色谱柱所得的液相色谱图。图10为采用合适的升温程序时的气相色谱图。图11为四个提取时间的液相图谱。图12为四个提取时间的气相图谱。图13为20批腰肾膏的液相指纹图谱。图14为20批腰肾膏的气相指纹图谱。具体实施方式本发明提供一种腰肾膏指纹图谱的建立方法及腰肾膏对照指纹图谱,为使本发明的目的、技术方案及效果更加清楚、明确,以下对本发明进一步详细说明。应当理解,此处所描述的具体实施例仅仅用以解释本发明,并不用于限定本发明。以下通过具体实施例对本发明做进一步说明。实施例中所涉及的仪器和试剂如下:1、仪器:安捷伦agilent1100高效液相;赛默飞thermou3000高效液相;安捷伦gc7890b;chromeleon色谱工作站;openlab2.0色谱工作站;中药指纹图谱色谱相似度评价系统(2012.130723版本);超声机:branson5510、昆山市超声仪器有限公司kq-400de;纯水机:密理博明澈-d24;天平:赛多利斯电子天平sqp;滤膜:津腾0.45μm微孔滤膜。2、试剂:色谱纯:甲醇、乙腈:fisherchemical;三氟乙酸:麦克林。分析纯:甲醇:广东光华科技股份有限公司;乙酸乙酯:广东光华科技股份有限公司、江苏强盛功能化学股份有限公司;无水硫酸钠:国药集团化学试剂有限公司。3、对照品:中国食品药品检定研究院:α-蒎烯110897-201803;薄荷脑110728-201707;桉油精110788-201707;丁香酚110725-201716;反式茴香脑111835-201503;龙脑110881-201709;桂皮醛110710-201821;蛇床子素110822-201710;补骨脂素110739-201617;中国药品生物制品检定所:异补骨脂素110738-200511。4、腰肾膏制剂国药集团德众(佛山)制药有限公司(批号:18046、18026、18013、18022、18031、18036、18002、18047、18008、18011、18052、18024、18032、19001等)实施例1:方法学验证选用批次为18046的腰肾膏六份,按拟定方法制备供试品溶液。取第一份做精密度和稳定性考察;总六份做重复性考察。供试品溶液的制备:取三片腰肾膏剪成条状,撕去背衬,以一片为单位结合成团,塞入250ml烧瓶中,加水100ml,接上载有2ml乙酸乙酯的挥发油提取器,在电热套上回流3小时;回流完毕后放冷,将挥发油提取器的乙酸乙酯层放入分液漏斗中,分别用乙酸乙酯2ml、2ml、1ml冲洗挥发油提取器,洗液并入分液漏斗中,弃去水层,乙酸乙酯层通过载有1g无水硫酸钠的滤纸,滤入10ml容量瓶中,加乙酸乙酯至刻度,摇匀,得到气相鉴别用供试品。提取后的膏体弃去水浸液,用100ml纯化水荡洗,重复三次,在105℃烘干,取其三分之一的重量,剪碎,置于50ml具塞锥形瓶中,加入50%甲醇30ml,超声60min,放冷,浸液用0.45μm微孔滤膜过滤,滤液作为液相鉴别用供试品。一、液相方法学验证色谱条件:仪器:thermou3000高效液相;色谱柱:welchultimatexb-c184.6*250mm,5μm;流动相:乙腈—0.02%三氟乙酸溶液;色谱柱:welchultimatexb-c184.6*250mm,5μm;波长:245nm;进样量20μl;洗脱条件如下表1所示。表1时间min乙腈(%)0.02%三氟乙酸溶液(%)015853547535547536550508065358773279710001191000指纹图谱评价软件参数:多点校正、时间窗0.3、中位数。1、精密度在以上拟定的实验条件基础上,将同一液相样品溶液重复进样6次,进样量20μl,记录液相指纹图谱,计算相互相似度,与其生成的对照指纹图谱相似度为1,共有峰保留时间rsd小于1%,说明该方法精密度良好。2、重复性在以上拟定的实验条件基础上,将同一样品分6份按液相样品方法进行提取,进样量20μl,记录液相指纹图谱,计算相互相似度,与其生成的对照指纹图谱相似度为1,共有峰保留时间rsd小于1%,说明该方法重复性良好。3、稳定性在以上拟定的实验条件基础上,将同一液相样品溶液,分别于不同时间(0h、12h、29h、41h)进样,进样量20μl,计算相互相似度,供试品溶液在41小时内,各时间点进样得到的色谱图相似度在0.99以上,共有峰保留时间rsd(%)小于1%,说明液相供试品溶液在41小时内稳定。4、色谱图记录时间延长一小时在以上拟定的实验条件基础上,选用液相样品溶液一批,进样20μl,将液相方法延长多1小时,得到的液相色谱图无后续出峰,说明仪器方法设定的时间内供试液成分可以完整出峰。二、气相方法学验证色谱条件:仪器:agilentgc7890b气相色谱仪;色谱柱:zb-wax;进样口:200℃;检测器:280℃;流速:1ml/min;进样量:1μl;升温速率如表2所示。表2速率℃/min温度℃保持时间min9071011510315000.516001024030指纹图谱评价软件参数:多点校正、时间窗0.3、中位数。1、精密度在以上拟定的实验条件基础上,将同一气相样品溶液重复进样6次,进样量:1μl,记录气相指纹图谱,计算相互相似度,进样6次与其生成的对照指纹图谱相似度为1,共有峰保留时间rsd(%)小于1%,说明该方法精密度良好。2、重复性在以上拟定的实验条件基础上,将同一样品分6份按气相样品方法进行提取,进样量:1μl,记录气相指纹图谱,计算相互相似度,提取6次与其生成的对照指纹图谱相似度为1,共有峰保留时间rsd(%)小于1%,说明该方法重复性良好。3、稳定性在以上拟定的实验条件基础上,将同一份气相样品溶液,分别于不同时间(0h、9h、17h、29h)进样,进样量:1μl,计算相互相似度,各时间点进样得到的色谱图相似度在0.99以上,共有峰保留时间rsd(%)小于1%,说明气相供试品溶液在29小时内稳定。4、色谱图记录时间延长一小时在以上拟定的实验条件基础上,选用气相样品溶液一批,将气相方法延长多1小时,得到的气相色谱没有后续出峰,说明仪器方法设定的时间内供试液成分可以完整出峰。实施例2:腰肾膏指纹图谱的建立一、排除辅料峰1、液相图谱辅料峰腰肾膏是由混合好的膏团涂布在弹性布上而制成的。弹性布在甲醇和乙醇的浸泡下,使弹性布上的染料被洗脱,进入供试品溶液中。为此,试分别使用50%乙醇和50%甲醇对腰肾膏的弹性布进行回流,减少被洗脱染料的量,以减少相似度评价的干扰。色谱条件:仪器:agilent1100高效液相色谱仪。洗脱条件如下表3所示。其他色谱条件与实施例1拟定的实验条件相同。表3时间min乙腈(%)0.02%三氟乙酸溶液(%)03070206040408020581000611000673070733070结果表明:使用50%甲醇进行提取得到的辅料峰较少,故选择本发明中采用50%甲醇进行提取。辅料峰在积分过程中剔除,不参与指纹图谱评价。2、气相图谱辅料峰将腰肾膏18013、弹性布辅料、空白组(空白组是不加入任何样品进行提取)按实施例1拟定的实验条件进行提取和测定,但升温速率与实施例1不同,如表4所示。表4速率℃/min温度℃保持时间min507101151571250315000.7165011700102400结果发现辅料萜烯树脂在气相色谱中有明显色谱峰,因此该色谱峰在不参与指纹图谱评价。二、液相指纹图谱选取20批腰肾膏按照实施例1拟定的方法进行提取和测定,记录指纹图谱,如图13所示。将指纹图谱导入中药色谱指纹图谱相相似度软件,采用平均值为对照图谱的生成方式,多点校正,时间窗宽度0.3,中位数,生成液相对照指纹图谱,如图1所示。计算20批腰肾膏样品与生成的对照指纹图谱相似度,20批样品相似度为0.912到0.990。三、气相指纹图谱选取20批腰肾膏按照实施例1拟定的方法进行提取和测定,记录指纹图谱,如图14所示。将指纹图谱导入中药色谱指纹图谱相相似度软件,采用平均值为对照图谱的生成方式,多点校正,时间窗宽度0.3,中位数,切除前4分钟的溶剂峰,生成气相对照指纹图谱,如图2所示。计算20批腰肾膏样品与生成的对照指纹图谱相似度,20批样品相似度为0.997到1。四、专属性1、液相指纹图谱特征峰指认对腰肾膏的液相对照指纹图谱的52个共有峰进行标号,如图2所示。(1)对照品特征峰使用五味子醇甲、补骨脂素、异补骨脂素、蛇床子素对腰肾膏指纹图谱进行归属。取五味子醇甲、补骨脂素、异补骨脂素及蛇床子素适量,加甲醇溶解,得对照溶液,按照实施条例1中的液相条件进行分析,并与腰肾膏供试品色谱图进行比较。结果如图3所示,其中,a为腰肾膏,b为五味子醇甲,c为补骨脂素、异补骨脂素,d为蛇床子素。五味子醇甲来源于五味子,补骨脂素及异补骨脂素来源于补骨脂,蛇床子素来源于蛇床子。三个药材为腰肾膏原料药材,因此上述三个峰可以体现为该药材的专属性峰,有助于腰肾膏质量的评价。(2)药材特征峰1)药材提取液与药材阴性溶液制备制备对照药材储备液:取药材适量,切碎,置250ml烧瓶中,加水75ml,按实施例1方法分别进行提取,得气相鉴别药材储备液和液相鉴别药材储备液。阴性药材溶液(液相):吸取除目标药材外的药材储备液各1ml,混合,加50%甲醇90ml,超声60min,即得。(3)药材专属峰1)枫香脂特征峰对腰肾膏的液相鉴别用供试液、枫香脂阴性溶液、枫香脂提取液按照实施例1拟定的色谱条件进行测定,确定12号,37号,38号峰为枫香脂特征峰。2)补骨脂特征峰对腰肾膏的液相鉴别用供试液、补骨脂阴性溶液、补骨脂提取液按照实施例1拟定的色谱条件进行测定,确定13号,14号,22号,26号,31号,42号峰为补骨脂特征峰。3)蛇床子特征峰对腰肾膏的液相鉴别用供试品、蛇床子阴性溶液、蛇床子提取液按照实施例1拟定的色谱条件进行测定,确定15号,21号,23号,27号,28号峰为蛇床子特征峰。4)五味子特征峰对腰肾膏的液相鉴别用供试品、五味子阴性溶液、五味子提取液按照实施例1拟定的色谱条件进行测定,确定20号,24号,25号,30号,39号,40号峰为五味子特征峰。5)乳香特征峰对腰肾膏的液相鉴别用供试品、乳香阴性溶液、乳香提取液按照实施例1拟定的色谱条件进行测定,确定49号峰为乳香特征峰。综上,腰肾膏液相指纹对照图谱中12号、13号、14号、15号、20号、21号、22号、23号、24号、25号、26号、27号、28号、30号、31号、37号、38号、39号、40号、42号、49号峰(共21个)为药材专属特征峰。2、气相指纹图谱特征峰指认对腰肾膏的气相对照指纹图谱的51个共有峰进行标号,如图1所示。(1)对照品特征峰按照实施例1拟定的色谱条件,对腰肾膏气相鉴别用供试液,对照品桉油精、薄荷酮、薄荷脑、龙脑、反式茴香脑、丁香酚的保留时间进行定位(桉油精、薄荷酮、薄荷脑均为薄荷素油的特征成分,龙脑是冰片的特征成分,丁香酚是丁香的特征成分,反式茴香脑是八角茴香和小茴香共有的特征成分),确定4号峰为桉油精,10号峰为薄荷酮,25号峰为薄荷脑,30号峰为龙脑,38号峰为反式茴香脑,46号峰为丁香酚。对腰肾膏气相鉴别用供试液,桂皮醛对照品的保留时间进行定位,确定44号峰为桂皮醛。对腰肾膏气相鉴别用供试液,樟脑、水杨酸甲酯对照品的保留时间进行定位,确定13号峰为樟脑,36号峰为水杨酸甲酯。对腰肾膏气相鉴别用供试液,薄荷素油对照品的保留时间进行定位,确定1号峰、2号峰、3号峰、4号峰、10号峰、11号峰、12号峰、16号峰、17号峰、25号峰、26号峰为薄荷素油特征峰。与上述对照品比较所得,其中4号峰为桉油精,10号峰为薄荷酮,25号峰为薄荷脑,三者均为薄荷素油的特征成分。按照实施例1拟定的色谱条件,对腰肾膏19010的气相鉴别用供试品,冰片对照品的保留时间进行定位。结果如图4所示,其中,a为腰肾膏19010,b为冰片。由图4可知,27号峰、30号峰为冰片特征峰。与上述对照品比较可知,30号峰为龙脑,是冰片的特征成分。按照实施例1拟定的色谱条件,对腰肾膏19010的气相鉴别用供试品,肉桂油对照品的保留时间进行定位。结果如图5所示,其中,a为腰肾膏19010,b为肉桂油。由图5可知,44号峰为肉桂油特征峰。与上述对照品比较可知,44号峰为桂皮醛,是肉桂油的特征成分。使用上述对照药材储备液制备方法制备得到的枫香脂药材储备液,吸取1ml,稀释10倍后,进行气相分析,见图6。按照实施例1拟定的色谱条件,对腰肾膏19010的气相鉴别用供试品,枫香脂药材储备液对照品的保留时间进行定位。结果如图6所示,其中,a为腰肾膏19010,b为枫香脂药材储备液。由图6可知,19号峰为枫香脂特征峰。使用上述对照药材储备液制备方法制备得到的丁香药材储备液,吸取1ml,稀释10倍后,进行气相分析,见图7。按照实施例1拟定的色谱条件,对腰肾膏19010的气相鉴别用供试品,丁香药材储备液对照品的保留时间进行定位。结果如图7所示,其中,a为腰肾膏19010,b为丁香药材储备液。由图7可知,46号峰,48号峰为丁香特征峰。与上述对照品比较所得,46号峰为丁香酚,是丁香的特征成分。使用上述对照药材储备液制备方法制备得到的八角茴香和小茴香药材储备液,吸取1ml,稀释10倍后,进入气相分析,见图8。按照实施例1拟定的色谱条件,对腰肾膏180401批的气相鉴别用供试品,八角茴香和小茴香药材储备液对照品的保留时间进行定位。结果如图8所示,其中,a为腰肾膏180401,b为八角茴香药材储备液,c为小茴香药材储备液。由图8可知,38峰为反式茴香脑,是八角茴香和小茴香共有的特征成分。因此,腰肾膏气相指纹对照图谱中1号、2号、3号、4号、10号、11号、12号、13号、16号、17号、19号、25号、26号、27号、30号、36号、38号、44号、46号、48号峰(共20个)为药材专属峰。对腰肾膏全面质量评价提供有利的参考。五、耐用性1、液相色谱条件(1)色谱柱的耐用性1)同一品牌色谱柱色谱柱:welchultimatexb-c184.6*250mm,5μm按照实施例1拟定的实验条件,以腰肾膏19010为样品,采用同一品牌的三根色谱柱进样,记录记录液相指纹图谱,如图9所示,其中,s1为色谱柱1,s2为色谱柱2,s3为色谱柱3。得到的液相色谱图导入中药指纹图谱色谱相似度评价软件,计算相互相似度,匹配结果如表5所示。同一品牌三根色谱柱相互相似度大于0.99,说明该品牌色谱柱重复性良好。表52)不同品牌色谱柱色谱柱:kromasil100-5c184.6*250mm,5μm;welchultimatexb-c184.6*250mm,5μm;thermoaypersilgolc184.6*250mm,5μm。按实施例1拟定的实验条件,以同一批液相样品溶液,采用不同品牌的三根色谱柱进样,记录液相指纹图谱,结果发现色谱柱局部出峰顺序或大小不一,宜选择同一品牌welchultimate色谱柱,以保证峰形和出峰时间与腰肾膏对照图谱接近。(2)柱温耐用性在实施例1拟定的实验条件基础上,以同一批液相样品溶液,采用不同的柱温(25℃、30℃、35℃)条件进样测试,结果显示柱温到达35℃,部分峰形发生变化,出峰时间偏移大,故液相指纹图谱适宜在25℃到30℃的柱温条件下分析。(3)不同高效液相在实施例1拟定的实验条件基础上,以同一批液相样品溶液,采用不同的高效液相色谱仪(agilent1100,thermou3000)进样测试,结果发现两台仪器测得液相指纹图谱相似度rsd小于0.2%,表明该分析方法用不同的高效液相色谱仪测定重复性良好。六、方法与结果(一)色谱条件选择1、液相方法仪器:agilent1100dad检测器;色谱柱welchultimatexb-c184.6*250mm,5μmagilent1100。1)波长选择选取一份液相样品溶液,设定扫描波长210nm-400nm,柱温30,进样量10μl,流动相甲醇(a)-水(b)梯度:0~20min,a:b=30:70;20~40min,a:b=60:40;40~58min,a:b=80:20;58~67min,a:b=100:0;67~73min,a:b=30:70。结果显示245nm色谱峰较多,峰大小较均衡,故选择245nm为检测波长。2)流动相选择采用实施例1拟定的试验条件,采用不同的流动相进行测定,结果如下:a)乙腈(a)-水(b)梯度:0~20min,a:b=10:90;20~40min,a:b=40:60;40~58min,a:b=80:20;58~67min,a:b=100:0;67~73min,a:b=90:10。b)甲醇(a)-0.1%磷酸溶液(b)梯度:0~20min,a:b=30:70;20~40min,a:b=60:40;40~58min,a:b=80:20;58~67min,a:b=100:0;67~73min,a:b=30:70。c)乙腈(a)-0.1%磷酸溶液(b)梯度:0~20min,a:b=10:90;20~40min,a:b=40:60;40~58min,a:b=80:20;58~67min,a:b=100:0;67~73min,a:b=90:10。d)乙腈(a)-0.1%磷酸溶液(b)梯度:0~20min,a:b=30:70;20~40min,a:b=60:40;40~58min,a:b=80:20;58~67min,a:b=100:0;67~73min,a:b=30:70。e)乙腈(a)-0.1%甲酸溶液(b)梯度:0~20min,a:b=10:90;20~40min,a:b=40:60;40~58min,a:b=80:20;58~67min,a:b=100:0;67~73min,a:b=90:10。f)乙腈(a)-0.1%醋酸溶液(b)梯度:0~12min,a:b=20:80;12~40min,a:b=30:70;40~60min,a:b=45:55;60~80min,a:b=55:45;80~85min,a:b=80:20;85~90min,a:b=90:10;90~94min,a:b=100:0;94~100min,a:b=20:80.以上流动相测试结果表明,有机相使用乙腈,峰型和分离度效果较好。g)乙腈-不同浓度三氟乙酸溶液选择a、乙腈(a)-0.1%三氟乙酸溶液(b)梯度:0~12min,a:b=20:80;12~40min,a:b=30:70;40~60min,a:b=45:55;60~80min,a:b=55:45;80~85min,a:b=80:20;85~90min,a:b=90:10;90~94min,a:b=100:0;94~100min,a:b=20:80。b、乙腈(a)-0.05%三氟乙酸溶液(b)梯度:0~15min,a:b=5:95;15~30min,a:b=25:75;30~35min,a:b=35:65;35~55min,a:b=44:56;55~61min,a:b=53:47;61~76min,a:b=60:40;76~91min,a:b=70:30;91~102min,a:b=100:0。c、乙腈(a)-0.02%三氟乙酸溶液(b)梯度:0~18min,a:b=15:85;18~38min,a:b=27:73;38~73min,a:b=35:65;73~93min,a:b=55:45;93~98min,a:b=73:27;98~104min,a:b=75:25;104~119min,a:b=100:0。结果表明,0.02%-0.1%三氟乙酸浓度的水相得到的峰形改善差不多,由于三氟乙酸酸性较强,遂选择0.02%作为水相的浓度。低于0.02%则起不到峰形改善的作用。3)不同种类的色谱柱比较采用实施例1拟定的试验条件,以腰肾膏18013为样品,使用agilentzorbaxsb-phengl4.6×250mm,5μm色谱柱进行对比实验,检测设备为agilent1100高效液相色谱仪,流动相乙腈(a)-0.1%磷酸溶液(b)梯度:0~12min,a:b=20:80;12~40min,a:b=30:70;40~60min,a:b=45:55;60~80min,a:b=55:45;80~85min,a:b=80:20;85~90min,a:b=90:10;90~94min,a:b=100:0;94~100min,a:b=20:80;结果表明苯基柱分离度不佳。4)不同柱长的色谱柱使用welchultimatexb-c184.6*150mm,5μm,150mm色谱柱进行对比实验,检测设备为agilent1100高效液相色谱仪,结果表明150mm色谱柱分离度不佳,故使用welchultimatexb-c184.6*250mm,5μm色谱柱分离效果更适合。(2)气相方法1)色谱柱选择色谱柱:hp-5,zb-wax,hp-ffap,db-1,agilent7890b气相色谱仪。按照实施例1拟定的实验条件,以腰肾膏18015为样品,采用以上不同型号的色谱柱进样,采用表6的升温程序,记录气相指纹图谱。结果表明:zb-wax分离度较好,而ffap主峰虽然分离度合适但是出峰时间慢(接近柱子使用温度上限)。表62)合适的升温程序的选择经过对升温程序的多次调整,最后得到如表2所示的合适升温参数,按照实施例1拟定的实验条件,以腰肾膏18009为样品,用作分析腰肾膏气相指纹图谱,结果如图10所示。2、样品前处理条件选择:(1)挥发油提取时间选择选择使用挥发油提取器分离出腰肾膏中的挥发性成分,提取时间分别按2h、3h、4h、5h做四份样品,用乙酸乙酯吸收挥发性成分,定容至10ml,得到气相供试品溶液。剩余膏体加入50%甲醇100ml回流1小时,放冷至室温,用0.45μm微孔滤膜过滤,得到液相供试品溶液。1)四个提取时间的液相方法测定检测仪器:aglient1100高效液相色谱仪,进样量:10μl,柱温:30℃,波长:274nm,流动相甲醇(a):0.1%磷酸(b)梯度:0~20min,a:b=30:70;20~40min,a:b=60:40;40~58min,a:b=80:20;58~67min,a:b=100:0;67~73min,a:b=30:70;结果如图11所示。2)四个提取时间的气相方法测定检测仪器:agilentgc7890b气相色谱仪,进样量1μl,升温程序如表6所示。结果如图12所示。结果表明:提取时间3h、4h、5h样品,液相图谱趋向一致;提取4h、5h的样品与提取2h、3h样品的气相图谱相比,部分峰有损失,故选择3小时作为提取时间。(2)挥发成分吸收溶剂的选择:1)环己烷和乙酸乙酯吸收效果比较在挥发油提取中,分别以环己烷、乙酸乙酯2ml作为挥发性成分吸收溶剂。以相同提取时间的气相色谱条件进行分析。结果表明:环己烷和乙酸乙酯的吸收效果大致一致,此处选择乙酸乙酯。(3)膏体提取方式选择1)提取溶剂的选择提取挥发油后的膏体两份,倾尽水分后烘干,分别采用50%甲醇、50%乙醇100ml回流提取,提取液蒸干,用甲醇溶解定容至10ml容量瓶,高效液相测定法测定,进样量10μl,柱温:30℃,波长:274nm,流动相甲醇(a):0.1%磷酸梯度(b):0~20min,a:b=30:70;20~40min,a:b=60:40;40~58min,a:b=80:20;58~67min,a:b=100:0;67~73min,a:b=30:70;,结果表明:使用50%甲醇进行提取得到色谱图峰高比较均衡,所以使用50%甲醇为提取溶剂。2)膏体提取方式考察膏体提取得到的供试品溶液中,曾经使用活性炭作为脱色剂。活性炭可以吸附一部分的染料,但是同时也会吸附其他成分,在脱色过程中有不稳定的影响,因此不进行脱色,在积分过程中剔除辅料峰,使其不参与指纹图谱评价。膏体在回流加热后冷却后有絮状沉淀析出,使指纹图谱重复性不佳,由此选择50%甲醇进行超声1小时,作为膏体提取方式。应当理解的是,本发明的应用不限于上述的举例,对本领域普通技术人员来说,可以根据上述说明加以改进或变换,所有这些改进和变换都应属于本发明所附权利要求的保护范围。当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1