使用标志物化合物相关的色谱特征和质谱特征共同鉴定植物性药材的方法与流程
相关申请的交叉引用本申请要求2018年5月2日提交的名称为“methodsforauthenticatingbotanicalsusingamarkercompound’srelatedchromatographicprofileandmassspectralprofilejointly(使用标志物化合物相关的色谱特征和质谱特征共同鉴定植物性药材的方法)”的美国临时专利申请号62/665,727的优先权和权益,该专利申请的全部内容全文以引用方式并入本文。本公开涉及使用标志物化合物相关的色谱特征和质谱特征共同鉴定植物性药材的方法。特别地,本发明的技术涉及使用标志物-指纹图谱定向的方法,该方法关注一个或多个标志物并且使用标志物相关的色谱特征和标志物相关的质谱特征来鉴定植物性药材或复杂样本。
背景技术:
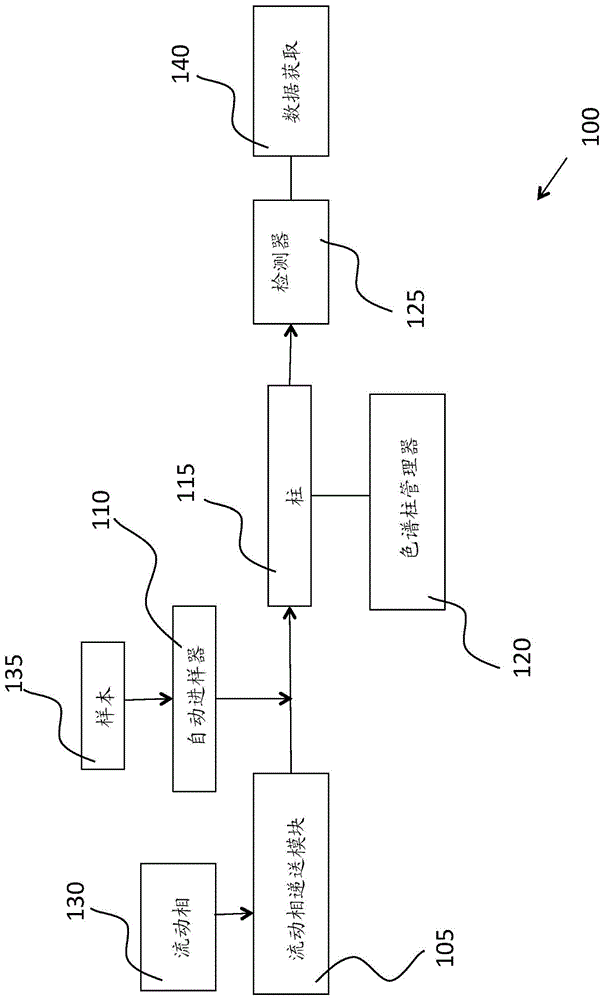
:植物性药材成分广泛用于饮食补充剂、草药、化妆品和个人护理产品中。由于缺乏生产标准化而导致的植物的潜在污染或错误识别已成为消费者的健康问题。探知植物性药材成分和加工产品的真实性是一项具有挑战性的任务,这是归因于复杂的植物化学组分、植物化学特征的自然变异以及密切相关的物种中类似的植物化学特征。液相色谱(lc)与质谱(ms)相结合是用于真实性评估的最有效的分析技术之一。然而,由于质谱仪的相对高成本和所需的高水平专业知识,lc-ms尚未广泛用于进行真实性测试的常规分析实验室。通常存在两种可用于植物性药材鉴定的技术。一种是标志物定向的方法,另一种是指纹图谱定向的方法。在标志物定向的方法中,一种或若干种化合物被识别为真实植物性药材的标志物。这些标志物化合物的存在或浓度水平被用作真实性评估的基础。在指纹图谱定向的方法中,收集真实植物性药材的一个或若干个指纹图谱并且在鉴定过程中将其用作一个或多个参考。指纹图谱可以是色谱特征或光谱特征,诸如uv/vis、ft-ir、nmr光谱。指纹图谱方法适用于复杂样本,尤其是当关键标志物或关键活性成分不能很好地确定时。例如,传统的草药通常通过指纹图谱定向的方法进行测试以便进行鉴定和/或质量控制。化学计量分析技术(诸如主组分分析、相似性分析、聚类分析)是也可用于植物性药材鉴定和分类的强大数据处理工具。然而,这些技术中的数据处理通常复杂且耗时。技术实现要素:所需要的是用于分析植物性药材和其它复杂化合物的耗时更少且更简单的方法,该方法可在常规分析实验室中进行而无需使用复杂的数据处理技术。本发明技术包括包括标志物定向的方法和指纹图谱定向的方法的组合。本发明技术关注一个或多个(例如,两个、三个、四个、五个等)标志物,并且使用标志物相关的色谱特征和标志物相关的质谱特征来鉴定植物性药材或复杂样本。例如,在下面的黑升麻实施例中,升麻酮醇3-o-α-l-拉伯糖苷用作目标标志物,并且其所提取离子色谱特征(其分子离子质荷比(m/z)处的)及其所提取质谱特征(标志物的色谱峰位置处的)一起用作黑升麻鉴定的基础。本发明技术的一个益处是,与指纹图谱定向的方法相比,处理数据简单得多。指纹图谱定向的方法通常使用复杂的数据分析工具,这些工具是耗时且复杂的。此外,通常需要特定数据处理软件。这不适用于常规分析实验室。本公开的技术,即标志物-指纹图谱方法,使用一种或多种标志物化合物的相关二维指纹图谱的特征来简化数据处理。数据处理简单、快速且易于在常规分析实验室中实现。在一方面,本发明技术的特征在于一种用于鉴定植物性药材的方法。该方法包括识别植物性药材的至少一种标志物化合物。将包括植物性药材的样本进样到色谱系统中,色谱系统包括流动相递送模块、自动进样器、色谱柱、色谱柱管理器和至少一个检测器。该方法还包括提取至少一种标志物化合物的色谱并且提取至少一种标志物化合物的色谱峰保留时间处的光谱。所提取色谱包括至少一个峰,并且所提取光谱包括至少一个谱带。对所提取色谱的至少一个峰进行积分。该方法还包括对所提取色谱的至少一个峰进行定量并且对所提取光谱的至少一个谱带进行定量。将所提取色谱的所定量至少一个峰和所提取光谱的所定量至少一个谱带与一组鉴定标准进行比较以确定植物性药材的真实性。该方法可包括以下实施方案或特征中的一者或多者。在一些实施方案中,植物性药材包括用于化妆品和个人护理产品的植物提取物、植物粉末、植物酊、草药或植物性药材成分。包括植物性药材的样本可具有约5mg/ml的一致浓度。在其它实施方案中,样本具有适合于所用检测器的一致浓度,例如,约1mg/ml、约2mg/ml、约3mg/ml、约4mg/ml、约6mg/lm、约7mg/ml、约8mg/ml、约9mg/ml或约10mg/ml。一致浓度允许一定的浓度变化,因为数据处理方法可被归一化。然而,如果浓度太低并且几乎没有检测到任何信号或者浓度太高而导致信号截断,则需要将浓度调整到适当的水平(即,在过低与过高情形之间)。一般来讲,优选制备具有相同浓度的样本。适当的质量检测器包括但不限于质量检测器(可从waterstechnologiescorporation,milford,ma商购获得)、sq检测器2(可从waterstechnologiescorporation,milford,ma商购获得)、tqd质量检测器(可从waterstechnologiescorporation,milford,ma商购获得)。在一些实施方案中,色谱系统是液相色谱系统、气相色谱系统或超临界流体色谱系统。检测器可为质谱分析仪。在一些实施方案中,质谱分析仪以扫描模式操作。可提取表征至少一种标志物化合物的离子的质荷比处的色谱。检测器可为紫外-可见光分光光度计或光电二极管阵列检测器。可提取表征至少一种标志物化合物的uv-vis波长处的色谱。在一些实施方案中,检测器是傅里叶变换红外光谱仪。可提取表征至少一种标志物化合物的ir波数处的色谱。在一些实施方案中,检测器是核磁共振光谱仪。可提取表征至少一种标志物化合物的nmr化学位移处的色谱。在一些实施方案中,提取至少一个峰的顶点处的光谱。可从所提取光谱中减去基线。在一些实施方案中,对所提取色谱的至少一个峰进行积分包括检测至少一个峰的基线的起点和终点、计算至少一个峰的面积和高度以及找到至少一个峰的顶点保留时间。在一些实施方案中,对所提取色谱上的峰进行定量可包括计算至少一个峰相对于所提取色谱上的所有检测到的峰的峰面积的总和的相对峰面积。该方法还可包括识别表征至少一种标志物化合物的离子的质荷比,并且计算至少一个谱带的强度。在一些实施方案中,该方法包括识别最大吸收波长并且计算至少一个谱带的强度。该方法还可包括识别最大吸收波数并且计算至少一个谱带的强度。在一些实施方案中,该方法包括识别光谱峰的化学位移并且计算至少一个谱带的强度。强度可为所提取光谱中最强离子峰的相对离子峰丰度。可对所提取色谱的所有峰进行定量。可对所提取光谱的所有谱带进行定量。在一些实施方案中,对所提取色谱的至少一个峰进行定量包括对峰保留时间、峰面积、峰相对面积、峰高度、峰相对高度、峰分辨率或它们的组合进行定量。在一些实施例中,对所提取光谱的至少一个谱带进行定量包括对质荷比、离子强度、相对强度、相对丰度、吸光度、透光率、波长、波数、化学位移、信号强度或它们的组合进行定量。当所定量至少一个峰和所定量至少一个谱带的所有值都在预先确定的阈值内并且所提取光谱匹配预先确定的光谱时,可鉴定植物性药材。在一些实施方案中,确定植物性药材的至少两种标志物化合物。在一些实施方案中,提取色谱包括提取至少两种标志物化合物中每一者的色谱。在一些实施方案中,提取光谱包括提取至少两种标志物化合物色谱峰保留时间中的每一者处的至少两种标志物化合物中每一者的光谱。附图说明图1是根据本发明技术的例示性实施方案的色谱系统的示意图。图2是根据本发明技术的例示性实施方案的用于鉴定植物性药材的方法的流程图。图3示出了根据本发明技术的例示性实施方案的标准品中的升麻酮醇3-o-α-l-拉伯糖苷(m/z621道尔顿)、北美黑升麻(na1-3)和亚洲黑升麻(a1-3)样本的所提取离子色谱。图4a示出了根据本发明技术的例示性实施方案的北美黑升麻ms谱库中的参考质谱(顶部光谱)中的标志物(升麻酮醇3-o-α-l-拉伯糖苷)峰的质谱。图4b示出了根据本发明技术的例示性实施方案的与北美黑升麻ms谱库中的参考质谱(图4a)中的一个相匹配的样本(底部光谱)中的标志物(升麻酮醇3-o-α-l-拉伯糖苷)峰的质谱。图5是根据本发明技术的例示性实施方案的流程图,其示出了针对黑升麻使用单个标志物的色谱图案及其质谱图案的真实性数据处理方案。图6是根据本发明技术的例示性实施方案的针对商业和自制样本的软件真实性测试报告的屏幕截图。图7a是根据本发明技术的例示性实施方案的质谱,其示出了锥孔电压(50v)对升麻酮醇3-o-α-l-拉伯糖苷质谱的影响。锥孔电压在光谱中示出。图7b是根据本发明技术的例示性实施方案的质谱,其示出了锥孔电压(30v)对升麻酮醇3-o-α-l-拉伯糖苷质谱的影响。锥孔电压在光谱中示出。图7c是根据本发明技术的例示性实施方案的质谱,其示出了锥孔电压(15v)对升麻酮醇3-o-α-l-拉伯糖苷质谱的影响。锥孔电压在光谱中示出。图7d是根据本发明技术的例示性实施方案的质谱,其示出了锥孔电压(10v)对升麻酮醇3-o-a-l-拉伯糖苷质谱的影响。锥孔电压在光谱中示出。图8a是根据本发明技术的例示性实施方案的标准品中的升麻素(307道尔顿)、北美黑升麻(na1-3)和亚洲黑升麻(a1-3)样本的所提取离子色谱。为清楚起见,仅示出了所关注的xic部分。图8b是根据本发明技术的例示性实施方案的标准品中的升麻酮醇3-o-α-l-拉伯糖苷(621道尔顿)、北美黑升麻(na1-3)和亚洲黑升麻(a1-3)样本的所提取离子色谱。为清楚起见,仅示出了所关注的xic部分。图8c是根据本发明技术的例示性实施方案的标准品中的黄肉楠碱(659道尔顿)、北美黑升麻(na1-3)和亚洲黑升麻(a1-3)样本的所提取离子色谱。为清楚起见,仅示出了所关注的xic部分。图8d是根据本发明技术的例示性实施方案的标准品中的27-脱氧升麻烃(661道尔顿)、北美黑升麻(na1-3)和亚洲黑升麻(a1-3)样本的所提取离子色谱。为清楚起见,仅示出了所关注的xic部分。图9a是根据本技术的示例性实施方案的色谱,其示出了北美黑升麻(na1)、亚洲黑升麻(a1)和四个未知的黑升麻样本(u1-4)的xic色谱。图9a中的标志物峰用星号(*)标记。提取621道尔顿处的xic。对于所有色谱,条件相同。系统可从waterstechnologiescorporation(milford,ma)商购获得。图9b是根据本技术的示例性实施方案的色谱,其示出了可与图9a的xic相比的北美黑升麻(na1)、亚洲黑升麻(a1)和四个未知的黑升麻样本(u1-4)的色谱。系统可从waterstechnologiescorporation(milford,ma)商购获得。具体实施方式本发明技术包括用于鉴定植物性药材的标志物定向的方法和指纹图谱定向的方法的组合。有待鉴定的植物性药材可为植物提取物、植物粉末、植物酊或草药。植物性药材可为化妆品和/或个人护理产品的成分。本发明技术关注一个或多个(例如,两个、三个、四个、五个等)标志物,并且使用标志物相关的色谱特征和标志物相关的质谱特征来鉴定植物性药材或复杂样本。本文所述的一种或多种方法可用于使用色谱系统鉴定植物性药材。图1是可在该方法中使用的色谱系统100的示意图。色谱系统包括流动相递送模块105、自动进样器110、色谱柱115、色谱柱管理器120和检测器125。色谱系统100中可包括多于一个色谱柱115。在一些实施方案中,色谱系统包括多于一个流动相递送模块105、多于一个自动进样器110、多于一个色谱柱管理器120和/或多于一个检测器125。流动相递送系统105将流动相从流动相贮存器130递送到色谱系统100。自动进样器110将样本从样本贮存器135递送到流动相流动流,之后使组合流动流进入色谱柱115。色谱柱管理器120可用于在系统100中的不同柱之间自动切换以加热和冷却一个或多个柱115,并且跟踪关于每个柱的信息,例如,在每个柱中使用哪些溶剂来防止不同溶剂之间的污染。色谱柱管理器120与至少一个色谱柱115连通。至少一个检测器125位于色谱柱115的下游。在色谱系统100中可使用多于一个检测器或多于一种类型的检测器。检测器可为例如质谱分析仪、紫外-可见光分光光度计、光电二极管阵列检测器、傅里叶变换红外光谱仪、核磁共振光谱仪、或它们的组合。例如,在一些实施方案中,可使用两个质谱分析仪。在其它实施方案中,可使用质谱分析仪和核磁共振光谱仪。也可使用多个检测器或多种类型的检测器的其它组合。质谱分析仪可以扫描模式操作。在一些实施方案中,检测器是单四极质谱仪、串联四极质谱仪和/或飞行时间质谱仪。检测器的电离模式可为例如电喷雾电离模式(esi)。根据有待鉴定的特定植物性药材,也可使用其它电离模式。数据获取模块140可与至少一个检测器125通信。数据获取模块140可用于例如采集/收集并分析从检测器125接收的数据。数据获取模块140可为安装有软件以收集并分析数据(例如,以执行积分)的计算机。色谱系统100可为如本领域技术人员已知的适用于鉴定特定植物性药材的任何类型的色谱系统。例如,色谱系统100可为液相色谱系统、气相色谱系统或超临界流体色谱系统。类似地,一个或多个色谱柱115可为液相色谱柱、气相色谱柱或超临界流体色谱柱。在色谱系统100中使用的流动相可根据所使用的色谱系统的类型以及所鉴定的特定植物性药材而变化。本领域的普通技术人员将理解针对有待鉴定的植物性药材如何选择色谱系统和流动相。例如,在超临界流体色谱系统中,流动相可为二氧化碳,并且在液相色谱系统中,流动相可为去离子水与甲酸和/或乙腈/甲醇与甲酸。图2是示出了本发明技术的方法的流程图200。用于鉴定植物性药材的方法使用色谱系统,例如图1的色谱系统100。参见图2,该方法包括识别有待鉴定的植物性药材的标志物化合物205。特定标志物化合物将根据被鉴定的植物性药材而变化。在一些实施方案中,可识别有待鉴定的植物性药材的多于一种标志物化合物,例如至少两种标志物化合物。例如,可识别任何单个植物性药材的两种、三种、四种、五种、六种、七种或更多种标志物化合物。所用的标志物化合物的数量可取决于例如真实植物性药材的复杂性以及任何已知的掺杂植物性药材的复杂性。一种或多种标志物化合物可选自例如植物性药材的治疗组分或植物性药材的其它生物活性组分、特征组分、主要组分、协同组分、相关组分、有毒组分和/或一般组分。植物性药材的一种或多种标志物化合物的选择是本领域技术人员已知的,并且可例如通过使用特定植物性药材的一种或多种已知标志物化合物的数据库来完成。在一些实施方案中,候选标志物化合物选自文献,或选自使用高分辨率质谱仪和化学计量分析的植物性药材的探索性研究。在某些实施方案中,然后可通过将真实样本色谱和不真实样本色谱进行比较来进一步选择候选标志物或将所选择的候选标志物缩小至一种或几种化合物。是否选择一种或多种标志物化合物取决于多个因素,诸如,可如何容易地将真实样本与不真实样本区分开来。任选地,也可识别205标志物化合物的特征或独特离子的质荷比(m/z)。可识别标志物化合物的特征离子的多于一个质荷比。如果使用多种标志物化合物,则可识别每种标志物化合物的至少一种特征离子的质荷比。标志物化合物的特征离子的质荷比是本领域技术人员已知的。该方法还可任选地包括识别至少一种标志物化合物210的最大吸收波长、最大吸收波数和/或化学位移或光谱峰。将包括植物性药材的样本进样到215色谱系统(例如,图1的色谱系统100)中。可通过以下方式来将样本进样到色谱系统中:将样本与流动相混合,之后进入色谱柱。在色谱柱上分离样本,并且样本的所分离化合物从色谱系统洗脱并进入至少一个检测器。样本内植物性药材的浓度可为例如约1mg/ml至约10mg/ml。在一些实施方案中,浓度为约5mg/ml。参见图2,提取220至少一种标志物化合物的色谱。所提取色谱包括至少一个峰。当在用于鉴定植物性药材的方法中使用多于一种标志物化合物时,可提取所用的每种标志物化合物的色谱。例如,当使用两种标志物化合物来鉴定植物性药材时,可提取两个色谱,每种标志物化合物一个色谱。提取色谱的位置取决于所用的检测器的类型。如果质谱分析仪被用作检测器,则提取表征至少一种标志物化合物的离子的质荷比处的色谱。如果使用紫外-可见光分光光度计或光电二极管阵列检测器,则提取表征至少一种标志物化合物的uv-vis波长处的色谱。如果使用傅里叶变换红外光谱仪作为检测器,则提取表征至少一种标志物化合物的ir波数处的色谱。如果使用核磁共振光谱仪作为检测器,则提取表征至少一种标志物化合物的nmr化学位移处的色谱。可使用不同类型的检测器的组合,例如可使用质谱分析仪和紫外-可见光分光光度计两者。在该示例中,可提取表征至少一种标志物化合物的离子的质荷比处的色谱,并且可提取表征至少一种标志物化合物的uv-vis波长处的色谱。可使用适用于鉴定特定植物性药材的检测器的任何组合。此外,提取225至少一种标志物化合物的色谱峰保留时间处的光谱。所提取光谱包括至少一个谱带。可在至少一个谱带的顶点处提取光谱。可从所提取光谱中减去基线。当在色谱系统中使用多个检测器时和/或当使用多种标志物化合物时,可提取一种或多种标志物化合物的多个光谱。在一些实施方案中,跨以峰顶点为中心的rt窗口提取光谱。rt窗口可介于峰积分基线的起点和终点之间。光谱可被提取为rt窗口中的组合光谱或平均光谱。当在用于鉴定植物性药材的方法中使用多于一种标志物化合物时,可提取标志物化合物色谱峰保留时间中的每一者处的每种标志物化合物的光谱。例如,当在用于鉴定植物性药材的方法中使用两种标志物化合物时,可提取两种标志物化合物色谱峰保留时间中的至少一者处的两种标志物化合物中每一者的光谱。对所提取色谱的至少一个峰进行积分230。对所提取色谱的至少一个峰进行积分可包括检测至少一个峰的基线的起点和终点。可计算至少一个峰的面积和高度,并且可找到至少一个峰的顶点保留时间。积分可经由存储在例如图1的色谱系统100的数据获取模块140上的软件来完成。参见图2,该方法还包括对所提取色谱235的至少一个峰进行定量并对所提取光谱240的至少一个谱带进行定量。对所提取色谱上的峰进行定量可包括计算至少一个峰相对于所提取色谱上的所有检测到的峰的峰面积的总和的相对峰面积。在一些实施方案中,对所提取色谱的至少一个峰进行定量包括对峰保留时间、峰面积、峰相对面积、峰高度、峰相对高度、峰分辨率或它们的组合进行定量。峰保留时间、峰面积、峰相对面积、峰高度、峰相对高度和峰分辨率都是所提取色谱的可定量特性。可将这些所测量的可定量特性与真实植物性药材的预先确定的定量特性进行比较,以确定样本是否含有真实或掺杂(不真实)植物性药材。例如,可针对所提取色谱的每个可定量特性或可定量特性的子集预先确定阈值,并且如果测量值在预先确定的值的极限内(例如,在某一标准偏差或百分比内),则样本含有真实植物性药材。在一些实施方案中,如果所测量的可定量值在预先确定的值的1%、5%或10%内,则样本含有真实植物性药材。预先确定的值、范围或阈值的确定可基于一个或多个真实植物性药材样本的可定量特性的统计分析。这些阈值可针对已知的不真实植物性药材样本进行测试,以确保所选取的阈值可准确地区分真实植物性药材样本和不真实植物性药材样本。对所提取光谱的至少一个谱带进行定量可包括对质荷比、离子强度、相对强度、相对丰度、吸光度、透光率、波长、波数、化学位移、信号强度或它们的组合进行定量。质荷比、离子强度、相对强度、相对丰度、吸光度、透光率、波长、波数、化学位移和信号强度都是所提取光谱的可定量特性。可将这些所测量的可定量特性与真实植物性药材的预先确定的定量特性进行比较,以确定样本是否含有真实或掺杂植物性药材。例如,可针对所提取光谱的每个可定量特性或可定量特性的子集预先确定阈值,并且如果测量值在预先确定的值的极限内(例如,在某一标准偏差或百分比内),则样本含有真实植物性药材。在一些实施方案中,如果所测量的可定量值在预先确定的值的1%、5%或10%内,则样本含有真实植物性药材。预先确定的值、范围或阈值的确定可基于真实植物性药材样本的可定量特性的统计分析。这些阈值可针对已知的不真实植物性药材样本进行测试,以确保所选取的阈值将准确地区分真实植物性药材样本和不真实植物性药材样本。在一些实施方案中,对所提取色谱的单个峰和所提取光谱的单个谱带进行定量。在一些实施方案中,对所提取色谱的所有峰和所提取光谱的所有谱带进行定量。在一些实施方案中,计算245所提取光谱的一个或多个谱带的强度。强度可为所提取光谱中最强离子峰(即,基离子峰)的相对离子峰丰度。将所提取色谱的所定量至少一个峰和所提取光谱的所定量至少一个谱带与一组鉴定标准进行比较250以确定植物性药材的真实性。当所定量至少一个峰和所定量至少一个谱带的所有值都在预先确定的阈值或范围内并且所提取光谱匹配预先确定的光谱时,可鉴定植物性药材。在一些实施方案中,预先确定的光谱是光谱库的一部分。预先确定的阈值可包括例如质荷比、离子强度、相对强度、相对丰度、吸光度、透光率、波长、波数、化学位移、信号强度、峰保留时间、峰面积、峰相对面积、峰高度、峰相对高度、峰分辨率或它们的组合的阈值。在一些实施方案中,该方法还包括基于对已知的真实植物性药材样本和非真实(或掺杂)植物性药材样本的结果的分析来建立一组鉴定标准。真实植物性药材样本可表示真实植物性药材的自然变异。自然变异包括植物性药材收获季节和时间、收获的地理位置、植物解剖结构以及用于真实样本的处理和制备技术的变化。非真实(或掺杂)植物性药材样本可为表示潜在的非真实或掺杂植物性药材样本的样本。当基于分析真实植物性药材样本和非真实植物性药材样本的结果来确定该组鉴定标准时,可随机化进样次序(即,样本队列中的进样次序)以消除与进样次序相关的可能的伪影。可通过对真实植物性药材样本和非真实植物性药材样本的多次分析来确定该组鉴定标准。例如,可基于真实植物性药材样本的至少两次重复进样和非真实植物性药材样本的至少两次重复进样来确定该组鉴定标准。在一些实施方案中,可基于真实植物性药材样本和非真实植物性药材样本的六次重复进样来确定该组鉴定标准。鉴定标准是基于色谱和光谱的可定量特性的。使用来自植物性药材的色谱和光谱的鉴定标准导致二维分析;一个维度来自色谱且一个维度来自光谱。在一些实施方案中,鉴定标准包括来自色谱的至少一个可定量特性(例如,峰保留时间、峰面积、峰相对面积、峰高度、峰相对高度、峰分辨率)和来自光谱的至少一个可定量特性(例如,质荷比、离子强度、相对强度、相对丰度、吸光度、透光率、波长、波数、化学位移、信号强度)。在一些实施方案中,鉴定标准包括来自色谱的至少两个、至少三个、至少四个、至少五个或至少六个可定量特性以及来自光谱的至少两个、至少三个、至少四个、至少五个或至少六个可定量特性。在一些实施方案中,用于色谱和光谱的鉴定标准的可定量特性的数量是相同的。在其它实施方案中,用于色谱和光谱的鉴定标准的可定量特性的数量是不同的。在一些实施方案中,鉴定标准包括色谱的所有可定量特性和光谱的所有可定量特性。用于鉴定植物性药材的可定量特性的具体数量和类型可取决于例如有待鉴定的植物性药材的复杂性。在一些实施方案中,仅单个色谱峰和单个光谱谱带的可定量特性用于鉴定。在其它实施方案中,两个或更多个(例如,两个、三个、四个、五个、六个、七个、八个、九个或十个)色谱峰和两个或更多个(例如,两个、三个、四个、五个、六个、七个、八个、九个或十个)光谱谱带用于鉴定。在一些实施方案中,用于鉴定的峰和谱带的数量是相同的,并且在其它实施方案中,用于鉴定的峰和谱带的数量是不同的。在一些实施方案中,仅某些峰或谱带用于鉴定。例如,仅两个、三个或四个最强峰或谱带用于鉴定。强峰可为例如具有最高峰面积、最高峰相对面积、最高峰高度、最高峰相对高度或最高峰分辨率的峰。强谱带可为例如具有最高离子强度、最高相对离子强度或最高相对丰度的谱带。用于强度的可定量特性可根据有待鉴定的具体植物性药材而变化。在一些实施方案中,用于鉴定的谱带是基离子谱带和接下来的四个最强谱带。基离子谱带和强谱带可包括分子离子、碎片离子、一个或多个加合离子和/或一个或多个同位素离子。鉴定标准还可包括光谱库匹配,例如含有植物性药材的样本的光谱匹配真实植物性药材的预先确定的光谱。真实植物性药材的光谱可存储在植物性药材库中。植物性药材库可存储在计算机上,例如存储在图1的数据获取模块140上。本文所述的技术使用与一种或多种标志物化合物相关的相关二维指纹图谱来鉴定植物性药材。使用标志物的指纹图谱可简化数据处理,并且允许简单、快速且流线型的数据处理。本文所述的一种或多种方法可在标准色谱数据软件中实现,并且适用于常规分析实验室。特定于标志物化合物的所提取离子色谱和特定于标志物化合物峰保留时间的所提取质谱的共同使用可用于鉴定植物性药材。该方法同时使用至少二维数据。离子色谱形成一个维度,并且质谱形成另一个维度。这与现有方法的不同之处在于,在本文所述的一种或多种方法中使用特征特性而不是标志物的特性。例如,在标志物定向的方法中,标志物的色谱峰特性(诸如峰面积、峰高度)用于定量。在上述标志物-指纹图谱方法中,不仅标志物的峰特性用于鉴定,而且在同一离子色谱种检测到的一个或多个其它峰特性也用于鉴定。在另一个示例中,在标志物定向的方法中,特定标志物的离子强度在ms扫描、sit或mrm模式下用于lc-ms定量。在上述标志物-指纹图谱方法中,不仅标志物的离子强度用于鉴定,而且质谱的特征(包括其它加合离子和碎片离子的离子强度)也用于鉴定。本发明技术不同于指纹图谱定向的方法,因为在本发明技术中仅使用一个或多个标志物相关的指纹图谱。在指纹图谱定向的方法中,使用整个样本的ft-ir、nmr或其它光谱。指纹图谱定向的方法的这些光谱并非特定于标志物化合物。示例实施例1样本由合作伙伴提供了三种真实北美黑升麻提取物(na1-3)、三种亚洲黑升麻(actaeacimicifuga)提取物(a1-3)和四种商业黑升麻样本(u1-4)。将这些提取物用70%甲醇稀释至约5mg/ml。从chromadex(irvine,ca)购买了四种标准品,即升麻素、升麻酮醇3-o-α-l-拉伯糖苷(cimigenol-3-alpha-l-arabinoside)、27-脱氧升麻烃(23-epi-26-deoxyactein)和黄肉楠碱。这些标准品在70%甲醇中以约5μg/ml制备。标准品的结构、cas登记号和单一同位素质量如下所示。通过分别以95:5和90:10的质量比混合北美黑升麻样本(na1)和亚洲黑升麻(a1)来制备自制黑升麻样本m-5和m-10。通过0.2微米ptfe膜过滤样本溶液,之后进行分析。lc条件在该实施例中使用的液相色谱条件在表1中示出。表1:lc条件表2示出了液相色谱系统的洗脱梯度。表2:lc洗脱梯度检测器、质量检测器(可从waterscorporation(milford,ma)商购获得)的条件在表3中示出。表3:险测器条件结果和讨论用于鉴定的定量参数。选择升麻酮醇3-o-α-l-拉伯糖苷作为真实黑升麻的标志物。从北美黑升麻样本提取的标志物的分子离子质荷比(m/z621道尔顿)处的离子色谱(xic)共享简单且一致的图案,该图案与来自亚洲黑升麻样本的xic明显不同(参见图3)。此外,所提取的标志物的峰保留时间(rt)5.77分钟处的质谱显示出特征模式(参见图4)。来自两个正交维度的这些图案或指纹图谱是该北美黑升麻鉴定方法的基础。色谱图案:北美黑升麻的xic中的共同特征是存在具有约相等峰高度的两个主峰,并且标志物的峰为它们中的一个(图3,na1-3)。使用标志峰的rt及其峰相对面积作为定量参数来表征该色谱图案。可使用另外的参数,但是这两个参数足以有效地区分北美黑升麻与亚洲黑升麻样本。质谱图案:使用从北美黑升麻样本提取的质谱中的前五种丰度离子来表征图案。这些离子包括分子离子(基峰,m/z621da)、碎片离子(m/z603da)、钠加合离子(m/z643da)和同位素离子(m/z622da、644da)。将这些离子的m/z和相对强度(相对于基峰或分子离子)值用作用于鉴定的定量参数。应当指出,这些质谱得自北美黑升麻样本,而不是得自升麻酮醇3-o-α-l-拉伯糖苷标准品。因此,除分子离子和碎片离子之外,还必须包括那些加合离子和同位素离子,以捕获色谱系统rt5.77分钟处的北美黑升麻样本的总体光谱图案。色谱系统可从waterstechnologiescorporation(milford,ma)商购获得。ms库匹配:将所提取质谱(rt5.77分钟处的)存储在定制的北美黑升麻ms库中,并用于未知黑升麻样本真实性测试中的ms库搜索。ms库匹配结果的屏幕截图在图4中示出。用于自动化分析的阈值为了确定北美黑升麻的那些鉴定参数的阈值,通过一式三份地测量三种北美黑升麻和三种亚洲黑升麻样本。以随机化方式测量这些样本以避免与进样次序相关的伪影。表4示出了这些鉴定参数的统计平均值、标准偏差(sd)和阈值。在表4中,将rt的上限和下限设定为rt平均值的±1%。对于相对峰面积(面积%),将极限设定在离平均值的sd的3倍处。对于预期质量相对强度,将下限设定为低于平均值的sd的3倍处。不存在预期质量相对强度的上限。这些阈值主要在sd的3倍处选取,以覆盖北美黑升麻的潜在宽变异。使用可从waterscorporation(milford,ma)商购获得的软件进行数据处理。创建方法集以实施鉴定过程。图5示出了由软件使用的数据处理流程图。表5示出了在软件方法中使用的软件功能。表4:北美黑升麻的色谱图案和质谱图案的特性以及鉴定参数中的阈值*:rt的±1%用于上限和下限**:不使用质语图案的上限表5:北美黑升麻鉴定标准和在软件中使用的相关软件功能和字段数据处理方法集商业和自制黑升麻样本的分析通过该方法测试四种商业黑升麻样本(u1-4)和两种自制黑升麻样本(m-5和m-10)。图6示出了这些样本的软件报告的屏幕截图。通过软件自动地以红色标记任何不符合鉴定标准的情况,并且在图6中以星号(*)示出。真实、不真实和受污染的黑升麻样本都被正确地确定。鉴定方法的益处化学计量分析是植物性药材的探索真实性研究中强大的工具。然而,在常规分析环境中,它们太复杂且难以实现。在此,展示了一种新型鉴定方法,在该方法中,使用标志物化合物的二维指纹图谱来鉴定北美黑升麻。由于仅处理标志物的色谱指纹图谱和质谱指纹图谱,因此需要处理的数据量相对较小,并且数据处置相对简单。整个数据处理可在可从waterscorporation商购获得的适用于常规分析实验室的软件中自动化。结论描述了用于北美黑升麻真实性的自动化二维指纹图谱分析的细节。标志物化合物的色谱指纹图谱和质谱指纹图谱中的关键特征或图案通过一组定量参数(诸如rt、峰相对面积、m/z和离子相对强度)来表征。确定北美黑升麻的这些参数的阈值,并且将其用于自动化软件数据处理。该系统方法能够区分北美黑升麻与亚洲黑升麻样本,并且以5重量%检测亚洲黑升麻污染。应当指出,由于方法开发中所使用的参考或训练样本的数量有限,因此可能需要进一步验证该黑升麻真实性方法。该系统方法的特征包括质量检测器的使用、用于鉴定的标志物的二维指纹图谱的使用以及由软件进行的整个数据处理的自动化。质量检测器是可负担、易于学习和使用的。自动化软件数据处理是快速且客观的。这些特征适用于常规真实性测试,其中分析时间和分析专业知识可能有限。这种新的系统方法可容易地在常规分析实验室中实现,以用于饮食补充剂、草药、化妆品和个人护理产品中的植物性药材成分和成品的鉴定或质量控制,以保护产品质量和安全性。实施例2质量检测器被专门设计用于提供色谱和非专家用户对质量数据的访问。该质量检测器呈现了将lc-ms带到常规分析实验室环境的实际解决方案。在该实施例中,使用质量检测器进行真实性测试的可行性在北美(na)黑升麻(actaearecemosa)的研究中展示。在该实施例中,使用由质量检测器收集的ms数据开发用于植物性药材鉴定的方法。在盲测中,用四种商业黑升麻样本评估该鉴定方法的准确性。ms优于蒸发光散射检测器(elsd)的优点也突出显示。在该实施例中,使用实施例1的样本、表1所示的lc条件、表2所示的lc洗脱梯度和表3所示的检测器条件。此外,表6示出elsd条件。表6:elsd条件增益250压力45.0psi漂移管温度55℃喷雾器在50%功率水平下加热数据速率10pps滤波时间常数正常系统方法优化质量检测器默认设置适用于许多应用。在该黑升麻研究中,为了获得最大分子离子强度,优化检测器仪器参数,诸如探针温度、毛细管电压和标准品质谱上的锥孔电压。图7示出了锥孔电压对升麻酮醇3-o-α-l-拉伯糖苷质谱的影响。选择并使用产生最大分子离子强度的检测器参数。用于真实黑升麻的标志物的选择真实黑升麻或北美黑升麻由actaearacemosa的根和根茎制成。多年以来,由于缺乏生产标准化,因此黑升麻植物的潜在污染或错误识别已成为健康问题。与使用黑升麻产品相关联的不利肝毒性事件已被报道,并且怀疑不真实黑升麻可能导致一些意外事件。污染或错误识别通常发生于中国式actaea物种,诸如a.heracleifolia、a.dahurica和a.cimicifuga,以及生长在与北美黑升麻相同的区域中的北美actaea物种,诸如a.pachypoda、a.rubra和a.podocarpa。黑升麻的许多化学组分已被用作用于真实性测试的生物标志物。在这些标志物中,在该系统研究中筛选最常见的标志物,诸如升麻素、升麻酮醇3-o-α-l-拉伯糖苷、黄肉楠碱和27-脱氧升麻烃,以获得有待与检测器一起使用的一种或多种合适的标志物(参见图8a至图8d)。升麻素、升麻酮醇3-o-α-l-拉伯糖苷和27-脱氧升麻烃的分子离子([m+h]+)和黄肉楠碱的脱水离子(m+h-h2o]+)是它们相应质谱(数据未示出)中的主离子(基峰)。使用这些基峰离子的质荷比(m/z)分别根据ms扫描数据提取离子色谱。并且所得的所提取离子色谱(xic)在图8a至图8d中示出。在图8a至图8d中的xic的检验之后,选择升麻酮醇3-o-α-l-拉伯糖苷(图8b中m/z621da,保留时间5.8分钟)作为北美黑升麻的标志物,因为与来自亚洲黑升麻的图案相比,它在北美黑升麻中显示出最简单且独特的色谱图案。真实性数据处理方案为了使真实性方法适用于常规分析,设计了新的真实性数据处理方案。该方案使用标志物的独特色谱和质谱图案或指纹图谱来评估样本真实性(图5)。该真实性数据处理方案在3软件程序中使用ms库匹配和其它现有功能来实现。不使用特定化学计量软件程序。在实施例1中讨论了如何在3软件中开发和实现该方案的细节。为了支持该真实性数据处理方案,在软件中创建黑升麻ms库。根据ms扫描数据提取(用基线减法)来自北美黑升麻样本的升麻酮醇3-o-α-l-拉伯糖苷质谱,并将其存储在该定制的北美黑升麻ms库中。软件ms库匹配功能用于检查样本的质谱是否与库中的任何参考质谱相匹配。3软件ms库可定制构建、导出和导入。图4示出了ms库匹配结果。uv/vis光谱也可用于真实性测试。然而,当与质谱相比时,uv/vis光谱通常特异性更低。在这种情况下,升麻酮醇3-o-α-l-拉伯糖苷是紫外线透明的。因此,使用uv/vis光谱库进行黑升麻真实性测试是不可用的。商业和自制黑升麻样本的分析通过该方法测试四种商业黑升麻样本(u1-4)和两种自制黑升麻样本(m-5和m-10)。在完成评估之后显示出商业样本的来源。表7为这些样本的真实性测试结果的汇总。正确地确定了真实北美黑升麻(u2和u3)和不真实黑升麻(u1和u4)样本。还正确检测了受污染样本,包括5重量%受污染的黑升麻样本(m-5)。表7:黑升麻样本真实性测试结果和样本的来源用于真实性测试的ms和elsd的比较蒸发光散射检测器(elsd)通常用于黑升麻识别。用于真实性测试的ms和elsd的比较是用北美黑升麻(na1)、亚洲黑升麻(a1)和4种未知黑升麻样本(参见图9a和图9b)进行的。这些色谱的lc条件相同。根据色谱(图9a),很容易辨别样本u2和u3是真实黑升麻,而样本u1和u4不是。相反,难以根据elsd色谱(图9b)辨别哪种未知样本是真实黑升麻。图9a和图9b清楚地示出,ms是elsd更好的区分复杂的植物性药材样本的技术。结论在该实施例中,使用h-class系统(购自waterstechnologiescorporation,milfordma)和质量检测器(waterstechnologiescorporation,milfordma)开发了用于黑升麻鉴定的方法。具体地,筛选了四种黑升麻组分:升麻素、升麻酮醇3-o-α-l-拉伯糖苷、27-脱氧升麻烃和黄肉楠碱,并且选择升麻酮醇3-o-α-l-拉伯糖苷作为标志物。使用适用于常规真实性测试的新的自动化数据处理方法。在该数据处理方案中,评估标志物的色谱和质谱数据。该真实性方法用四种商业黑升麻样本和两种自制的受污染的北美黑升麻样本测试。正确地识别了所有真实和不真实或受污染样本。应当指出,由于方法开发中所使用的训练或参考样本的数量有限,因此可能需要进一步验证该黑升麻真实性方法。该实施例表明,质量检测器符合植物性药材鉴定,这是产品安全性和质量的基本测试。由质量检测器收集的升麻酮醇3-o-α-l-拉伯糖苷离子色谱具有较少的干扰峰,并且显示出北美黑升麻样本与亚洲黑升麻样本之间的明显差异。升麻酮醇3-o-α-l-拉伯糖苷质谱含有用于真实性测试的特征质谱图案。质量检测器使用简单且可负担,并且可广泛实施于用于植物性药材鉴定的常规分析实验室中。质量检测器在真实性测试中的采用可极大地提高植物性药材鉴定中的速度和置信度,并且有助于确保市场上的饮食补充剂、草药、化妆品和个人护理产品安全且质量高。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1