检测、预防、逆转和治疗神经系统疾病的方法
检测、预防、逆转和治疗神经系统疾病的方法
1.相关申请的交叉引用
2.本技术要求2018年12月5日提交的美国临时申请序列号62/775,626的优先权,其通过引用以其整体并入本文。
3.关于联邦资助的研究或开发的声明
4.不适用。
5.通过引用并入的材料
6.不适用。
技术领域
7.本公开总体上涉及神经系统疾病(例如,成人发作的神经系统疾病、阿尔茨海默病(ad)、帕金森病(pd)、额颞叶痴呆(ftd)等)的治疗和检测。
技术实现要素:
8.在本公开的各个方面中,提供了一种检测、预防、治疗、逆转或延迟神经系统疾病(例如,成人发作的神经系统疾病、ad、pd或ftd)的发作的方法。
9.本发明的一个方面提供了一种在对于包含功能丧失变体的溶酶体基因杂合的受试者中调节自噬
‑
溶酶体途径的方法,该方法包含:向需要其的受试者施用治疗有效量的自噬
‑
溶酶体途径调节剂。
10.本发明的一个方面提供了一种预防、治疗、逆转或延迟受试者中与溶酶体功能障碍相关联的神经系统疾病、障碍或病症的发作的方法,该方法包含:在受试者的生物样品中检测或已经检测到包含功能丧失变体的至少一种溶酶体基因;以及施用治疗有效量的自噬
‑
溶酶体途径调节剂,其中受试者对于包含功能丧失变体的溶酶体基因是杂合的或受试者是溶酶体贮积病(lsd)的携带者。
11.本发明的一个方面提供了检测受试者中至少一种溶酶体基因功能丧失变体的方法,包含:提供来自受试者的生物样品;以及检测包含功能丧失变体的溶酶体基因的存在,其中如果检测到至少一种包含功能丧失变体的溶酶体基因,则确定受试者具有与app加工功能障碍相关联的神经系统或神经退行性疾病、障碍或病症或处于与app加工功能障碍相关联的神经系统或神经退行性疾病、障碍或病症的风险中。在一些实施例中,该方法包含施用治疗有效量的自噬
‑
溶酶体途径调节剂,其中自噬
‑
溶酶体途径调节剂是与检测到的包含功能丧失变体的溶酶体基因相关联的治疗。
12.在一些实施例中,包含功能丧失变体的溶酶体基因与溶酶体贮积病(lsd)相关联。
13.在一些实施例中,受试者被怀疑患有与溶酶体功能障碍相关联的神经系统或神经退行性疾病、障碍或病症或处于所述与溶酶体功能障碍相关联的神经系统或神经退行性疾病、障碍或病症的风险中。
14.在一些实施例中,受试者对于溶酶体基因功能丧失变体是杂合的或是溶酶体贮积病(lsd)的携带者,或者被怀疑对于溶酶体基因功能丧失变体是杂合的或被怀疑是溶酶体
贮积病(lsd)的携带者。
15.在一些实施例中,包含功能丧失变体的溶酶体基因选自由以下组成的群组:ctns、man2b1、mfsd8、glb1、galns、naglu、cln3、gnptab、sgsh、cln8、npc1、tpp1、dnajc5、manba、ppt1、smpd1、gaa、hgsnat、gns、ctsa、hexb及其组合。
16.在一些实施例中,包含功能丧失变体的溶酶体基因与溶酶体贮积病(lsd)相关联,选自由以下组成的群组:aga、arsa、arsb、asah1、cln2(tpp1)、cln3、cln5、cln6、cln8、ctns、ctsa、ctsd、ctsk、fuca1、gaa、galc、galns、gla、glb1、gm2a、gnptab、gnptg、gns、gusb、hexa、hexb、hgsnat、hyal1、ids、idua、kctd7、lamp2、lipa、man2b1、manba、mcoln1、mfsd8、naga、naglu、neu1、npc1、npc2、ppt1、psap、sgsh、slc17a5、smpd1、sumf1、chit1、atp13a2、ctsf、dnajc5、grn及其组合。
17.在一些实施例中,包含功能丧失变体的溶酶体基因选自由以下组成的群组:neu1、naglu、gba、glb1、manba、man2b1、hgsnat、ids、ppt1、gns及其组合。
18.在一些实施例中,包含功能丧失变体的溶酶体基因选自由galc、acd及其组合组成的群组。
19.在一些实施例中,自噬
‑
溶酶体途径调节剂包含基因疗法(gt),其中gt增加或增强与包含功能丧失变体的溶酶体基因相关联的酶活性,包含功能丧失变体的溶酶体基因选自由以下组成的群组:aga、arsa、arsb、asah1、cln2(tpp1)、cln3、cln5、cln6、cln8、ctns、ctsa、ctsd、ctsk、fuca1、gaa、galc、galns、gla、glb1、gm2a、gnptab、gnptg、gns、gusb、hexa、hexb、hgsnat、hyal1、ids、idua、kctd7、lamp2、lipa、man2b1、manba、mcoln1、mfsd8、naga、naglu、neu1、npc1、npc2、ppt1、psap、sgsh、slc17a5、smpd1、sumf1、chit1、atp13a2、ctsf、dnajc5、grn及其组合。
20.在一些实施例中,用调节下列表达的自噬
‑
溶酶体途径调节剂治疗受试者:aga、arsa、arsb、asah1、cln2(tpp1)、cln3、cln5、cln6、cln8、ctns、ctsa、ctsd、ctsk、fuca1、gaa、galc、galns、gla、glb1、gm2a、gnptab、gnptg、gns、gusb、hexa、hexb、hgsnat、hyal1、ids、idua、kctd7、lamp2、lipa、man2b1、manba、mcoln1、mfsd8、naga、naglu、neu1、npc1、npc2、ppt1、psap、sgsh、slc17a5、smpd1、sumf1、chit1、atp13a2、ctsf、dnajc5、grn及其组合。
21.在一些实施例中,受试者对于包含功能丧失变体的溶酶体基因中的杂合变体是单倍不足的,或者在包含功能丧失变体的溶酶体基因中具有杂合变体,包含功能丧失变体的溶酶体基因选自由以下组成的群组:aga、arsa、arsb、asah1、cln2(tpp1)、cln3、cln5、cln6、cln8、ctns、ctsa、ctsd、ctsk、fuca1、gaa、galc、galns、gla、glb1、gm2a、gnptab、gnptg、gns、gusb、hexa、hexb、hgsnat、hyal1、ids、idua、kctd7、lamp2、lipa、man2b1、manba、mcoln1、mfsd8、naga、naglu、neu1、npc1、npc2、ppt1、psap、sgsh、slc17a5、smpd1、sumf1、chit1、atp13a2、ctsf、dnajc5、grn及其组合。
22.在一些实施例中,包含功能丧失变体的溶酶体基因选自由以下组成的群组中的负责硫酸乙酰肝素(hs)代谢的基因中的罕见功能变体:sgsh、naglu、hgsnat、gns及其组合。
23.在一些实施例中,功能丧失变体是选自由溶酶体基因的缺失、取代或缺失组成的群组的一种或多种变体。
24.在一些实施例中,神经系统或神经退行性疾病、病症或病症与溶酶体功能障碍相关联。
25.在一些实施例中,神经系统或神经退行性疾病、障碍或病症与改变的app加工(例如,改变的脑间质aβ水平和增加的aβ斑块负荷)或α
‑
syn聚集相关联。
26.在一些实施例中,神经系统或神经退行性疾病、障碍或病症与aβ积累相关联。
27.在一些实施例中,神经系统或神经退行性疾病、障碍或病症是阿尔茨海默病(ad)。
28.在一些实施例中,自噬
‑
溶酶体途径调节剂是与包含功能丧失变体的溶酶体基因相关联的药剂,包含选自由以下组成的群组:化学伴侣疗法(cct)、酶替代疗法(ert)、基因疗法(gt)、基因编辑、造血干细胞移植(hsct)、化学伴侣疗法(cct)、终止密码子通读药物、底物减少疗法(srt)及其组合。
29.在一些实施例中,方法包含通过酶替代疗法(ert)、基因疗法(gt)或干细胞疗法补充外源性溶酶体蛋白。
30.在一些实施例中,自噬
‑
溶酶体途径调节剂是与包含功能丧失变体的溶酶体基因相关联的治疗,该治疗包含半胱胺、环糊精或美格鲁特。
31.在一些实施例中,与未治疗的受试者相比,在受试者中aβ、apoe、tau或α
‑
syn聚集减少,或者aβ、apoe、tau或α
‑
syn清除增强。
32.在一些实施例中,受试者患有或被怀疑患有痴呆、阿尔茨海默病(ad)、帕金森病(pd)、额颞叶痴呆(ftd)、克雅氏病、运动神经元病、多聚谷氨酰胺障碍、亨廷顿病、家族性淀粉样多发性神经病(fap)、路易体痴呆或多系统萎缩。
33.其他目的和特征在下文中将部分地明显并且被部分地指出。
附图说明
34.本领域技术人员将理解,以下描述的附图仅用于说明目的。附图并不旨在以任何方式限制本教导的范围。
35.图1a至图1c是描绘naglu转录水平随(a)年龄、(b)阿尔茨海默病(ad)状态和(c)在ad小鼠模型中变化的一系列图。
36.图2是显示与健康对照相比,来自帕金森病(pd)病例的黑质的神经元中的naglu转录水平的点图。
37.图3a至图3b是一系列图像和蛋白免疫印迹(western blot),其描绘了在用预形成的α
‑
syn原纤维(pff)处理的神经元中α
‑
突触核蛋白(α
‑
syn)的积累。(a)在原代神经元中由α
‑
syn pff诱导的磷酸化的α
‑
syn聚集体的免疫荧光检测。用磷酸盐缓冲盐水(pbs)处理的神经元培养物被作为对照示出。(b)蛋白免疫印迹分析显示,在用1%triton x
‑
100并且然后用2%十二烷基硫酸钠(sds)连续提取(仅显示sds可溶部分)后,在来自pff处理的原代神经元的裂解物中积累了α
‑
syn。
38.图4a至图4c是显示来自不同基因型的小鼠的脑切片中磷酸化的α
‑
syn(psyn)染色的一系列图像。(a)接种(dpi)30天后用磷酸盐缓冲盐水(pbs)对年轻野生型(wt)c57bl/6小鼠进行psyn染色(棕色)和甲酚紫复染的冠状脑切片。(b)用α
‑
syn pff对30dpi的年轻wt c57bl/6小鼠进行psyn染色。(c)用α
‑
syn pff对30dpi的年轻naglu缺陷小鼠进行psyn染色。黑色箭头表示注射部位的水平。插图显示用抗psyn抗体染色和皮质中的路易体(lb)/路易神经突(ln)样病变。蓝色框位于外嗅皮层中。聚集体更为广泛(蓝色框),并且在naglu缺陷小鼠的同侧和对侧半球中均存在。比例尺:100μm。
39.图5是自噬
‑
溶酶体途径(alp)的降解能力的年龄依赖性下降及其在神经退行性变中的作用的示意图。黑线表示alp功能的年龄依赖性下降。蓝线表示阿尔茨海默病(ad)的神经退行性改变。虚线表示95%的置信区间(ci)。
40.图6是散点图,其比较了ad组群中溶酶体贮积病(lsd)致病基因的预测的功能性罕见变体的积累等位基因频率(cmaf)与外显子组聚合联盟(exome aggregation consortium)(exac)。从欧洲裔的ad组群中获得的每个lsd致病基因的积累等位基因频率位于x轴,并且exac数据集的积累等位基因频率位于y轴。
41.图7a至图7e是描绘溶酶体蛋白cspα在溶酶体功能中的作用的一系列图像和条形图。(a)n2a细胞(永生化神经元细胞系)的免疫染色的代表性图片。(b)n2a细胞的胞质和溶酶体富集部分的lamp
‑
1和cspα的代表性蛋白免疫印迹。(c)n2a细胞中的溶酶体追踪信号,该n2a细胞稳定地表达无(nothing)(空)、hcspα
‑
wt(野生型)或cspα
‑
p.l115r(p.l115r,ancl成人发作性神经元蜡样脂褐质病中的致病突变)。(d)图显示在稳定表达无(空)、hcspα
‑
wt(wt)或hcspα
‑
p.l115r(p.l115r)的细胞的细胞匀浆中测量的溶酶体酶ppt
‑
1、β
‑
gluc和β
‑
hexa的活性。(e)图显示在培养基(分泌型)中测量的ppt
‑
1、β
‑
gluc和β
‑
hexa的溶酶体酶活性。
42.图8a至图8c是显示dnajc5转录水平随年龄、ad状态和在小鼠模型中ad的变化的一系列点和线图(注意:dnajc5是编码溶酶体蛋白cspα的基因名称)。(a)在不同年龄时神经病理学正常的人脑样品中的dnajc5转录水平。(b)在两项不同的研究中,来自ad病例的神经元中的dnajc5转录水平与对照组的比较。(c)从2至18个月的在ad小鼠模型的皮层中dnajc5转录水平(tau,p.p301l;蓝线);(app,p.k670n/p.m671l;红线)和野生型小鼠(黑线)。右侧的图表示从2至18个月的相对斑块密度(app小鼠)和缠结密度(tau小鼠)。
43.图9a至图9d是显示cspα在体内和体外影响app处理的一系列图像和条形图。(a)成人发作lsd患者(ancl)的大脑皮层的app/aβ(4g8)染色的代表性图像。(b)用空载体、特异性shrna和引起ancl突变的p.l115r(p.l115r)转导的n2a细胞中app/aβ(4g8,红色)与lamp1(绿色)的代表性重叠共焦图像。(c)来自如(b)中转导的n2a细胞的条件培养基(培养基)和细胞匀浆(细胞)中的aβ40和aβ42水平。(d)上覆培养基中的可溶性appα片段、全长app及其α
‑
ctf和β
‑
ctf以及如(b)中所示转导的n2a细胞中的cspα的免疫印迹(左)和定量(右)。
44.图10a至图10b是显示溶酶体蛋白、npc1转录水平随(a)年龄和(b)ad状态变化的一系列点图。
45.图12a至图12d是显示内源性cspα位于溶酶体且突变体cspα影响alp功能的一系列图像、点图和条形图。(a)在原发性皮质神经元的体细胞和神经突以及神经元样细胞类型(n2a)中与溶酶体标记物共定位的内源性cspα的代表性图像和定量。(b)显示cspα与溶酶体标记物lamp1的共沉淀的蛋白免疫印迹。(c)用于表达成人发作性蜡样脂褐质病(ancl)致突变p.l115r的神经元中的溶酶体标记物的蛋白免疫印迹。(d)野生型和p.l115r细胞中的溶酶体追踪信号的定量。
46.图13a至图13b是显示cspα影响体内aβ产生的一系列图像和条形图。(a)来自ancl、对照和ad患者的脑切片的图像。(b)对来自ancl、ad和健康对照样品的脑样品的洗涤剂可溶和不可溶(胍)部分中的aβ的定量。
47.图14a至图14b是一系列条形图,其显示β淀粉样蛋白积累在对于ppt和naglu是半
合子的小鼠中被加剧。图14a半合子幼稚naglu小鼠在脑间质液(isf)中表现出较低的aβ水平。对同窝wt和半合子小鼠中aβ的isf水平进行的微透析定量揭示了aβ的基线isf水平显著降低。通过不成对的t检验,*p<0.05。10个月大的野生型(n=8)和naglu半合子(n=5)。图14b半合子幼稚ppt1小鼠在脑间质液(isf)中表现出较低的aβ水平。对同窝wt和半合子小鼠中aβ的isf水平进行的微透析定量揭示了aβ的基线isf水平显著降低。通过不成对的t检验,**p<0.01。7个月大的野生型(n=5)和naglu半合子(n=6)。
48.图15是显示在注射α
‑
syn预制原纤维(pff)后α
‑
syn病理的扩散的一系列图像。在三个月大的野生型小鼠(红色箭头)的纹状体内注射单剂α
‑
syn pff,并在注射后90天取出大脑,并用磷酸特异性α
‑
syn抗体染色。插图显示在多个大脑区域中的α
‑
syn病理,可见为棕色沉积物(甲酚紫复染剂以蓝色示出)。
49.图16a至图16b是显示在naglu缺陷小鼠中psyn聚集体增加的一系列图像。(a)在海马结构(上图)和黑质(下图)注射(dpi)α
‑
syn pff 90天后,对年轻野生型c57bl/6小鼠进行psyn染色(棕色)和甲酚紫复染的冠状脑切片。(b)在海马结构(上图)和黑质(下图)用α
‑
syn pff进行90dpi的年轻naglu缺陷小鼠的psyn染色。黑色箭头表示在naglu缺陷小鼠中的同侧和对侧半球中聚集体更广泛的脑区域。
50.图17a至图17c是一系列图像,其显示psyn染色(棕色)的黑质(sn)处的冠状脑切片,并用纹状体中具有α
‑
syn pff的年轻野生型c57bl/6小鼠90dpi的甲酚紫复染。(b)用纹状体中的α
‑
syn pff对年轻naglu缺陷小鼠90dpi的sn的psyn染色。(c)酪氨酸羟化酶(th,绿色)和psyn(红色)在年轻野生型小鼠90dpi的sn处与纹状体中的α
‑
syn pff共染色。
51.图18是使用表达相应溶酶体酶cdna的重组腺相关病毒(aav)载体,在ad(5xfad+/
‑
)的小鼠模型中治疗ad病理学的体内方法的示意图,该ad(5xfad+/
‑
)的小鼠模型在溶酶体酶基因中含有杂合突变(5xfad+/
‑
、naglu+/
‑
或5xfad+/
‑
、ppt1+/
‑
)。
52.图19是使用表达相应溶酶体酶cdna的重组aav载体在pd的小鼠模型(注射α
‑
syn预制原纤维)中治疗pd病理的体内方法的示意图,该pd小鼠模型在溶酶体酶基因(naglu+/
‑
或ppt1+/
‑
)中含有杂合突变。
53.图20显示与对照相比,来自帕金森病病例的黑质的神经元中的ppt1转录水平类似于图2的naglu转录水平。
54.图21a至图21f是一系列图像和图,其显示ad模型(5xfad+/
‑
)中的naglu、ppt1和dnajc5半合子会加剧β淀粉样蛋白积累。7个月大时(a)5xfad+/
‑
、(b)5xfad+/
‑
/naglu+/
‑
、(c)5xfad+/
‑
/ppt+/
‑
和(d)5xfad+/
‑
/dnajc5+/
‑
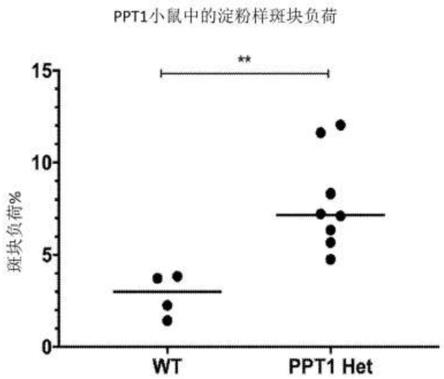
海马区域中的β淀粉样蛋白染色的代表性图片。图21e为点图,其显示ppt1基因的半合子加剧了β淀粉样蛋白的积累。与5xfad小鼠相比,在ppt1半合子/5xfad小鼠中由斑块所覆盖的表面积增加。海马结构中斑块负荷的量化被盲法地进行。通过不成对的t检验,**p<0.01。7个月大的5xfad(n=4)和ppt1半合子/5xfad(n=8)。图21f为点图,其显示naglu基因中的半合子加剧了β淀粉样蛋白的积累。与5xfad小鼠相比,在naglu半合子/5xfad小鼠中由斑块所覆盖的表面积增加。海马结构中的斑块负荷的量化被盲法地进行。通过不成对的t检验,*p<0.05。7个月大的5xfad(n=4)和naglu半合子/5xfad(n=4)。
55.图22是显示溶酶体基因调节α
‑
突触核蛋白聚集的功能丧失(lof)的一系列图像。naglu缺乏会加剧内源性小鼠α
‑
syn的病理性聚集。ppt1
‑
缺乏“减少”内源性小鼠α
‑
syn的病
理聚集。α
‑
syn预制原纤维的纹状体内接种注射后90天。
56.图23是显示靶向的aav2/9
‑
ppt1基因疗法延迟ppt1半合子/5xfad小鼠中疾病进展的存活曲线。kaplan
‑
meier生存曲线显示,未治疗的ppt1半合子/5xfad(红色,n=8)的中位生存期明显短于经颅内注射aav9
‑
ppt1治疗的半合子/5xfad(绿色,n=5)。通过对数秩检验进行趋势的分析对总生存期有显著意义(p<0.0001)。用aav2/9
‑
ppt1治疗的小鼠在第7.8个月时被特意取下用于组织学分析。
57.图24是描述患有婴儿神经元性蜡样脂褐质病cln1疾病的儿童的母亲和父亲(均为携带者)的谱系的图。
具体实施方式
58.本公开至少部分地基于以下发现:对于引起溶酶体贮积病(lsd)的溶酶体蛋白基因中有害突变杂合(携带者)的受试者(lsd的携带者,没有lsd症状)具有增加的发展阿尔茨海默病(ad)的风险。目前的教条是,导致lsd的基因中lof变体的杂合携带者和许多其他遗传性疾病被认为是正常的,并且不容易患任何疾病。
59.患有lsd的患者对于导致lsd的基因缺陷是纯合的(两个等位基因具有完全或几乎完全功能丧失的溶酶体基因变体),并且在婴儿期至20岁之间生活在任何地方,这取决于lsd。这些lsd患者不会活得足够长以发展ad。
60.如本文所述,本发明人发现,在患有ad的受试者中,检测并富集了引起lsd的基因中的一种或多种变体(有害突变),并且仅在一个等位基因(杂合的)中检测到。本发明人发现这些变体是杂合受试者中的功能丧失变体。
61.目前有多种治疗策略用于治疗溶酶体贮积病(lsd)(例如,酶替代疗法(ert)、基因疗法(gt)、干细胞疗法(sct)(例如,造血干细胞移植)、口服小分子底物减少疗法、小分子伴侣或自噬
‑
溶酶体途径的药理学补救),并且用于治疗具有溶酶体蛋白中有害杂合突变的ad患者。
62.溶酶体基因中杂合的功能丧失变体
63.众所周知,对于与lsd相关联的基因中功能丧失变体(例如,又称有害变体或突变)纯合的受试者会导致严重的溶酶体功能障碍。然而,本文令人惊讶地发现,人类ad患者的基因组富含溶酶体基因(例如lsd携带者)中的杂合完全的或几乎完全的功能丧失变体。此外,本文还表明,完全正常外观小鼠的溶酶体基因中有害突变的杂合性会导致微妙的溶酶体功能障碍,并直接影响正常的app加工。
64.如本文所述,功能丧失变体可以是完全功能丧失变体、几乎完全功能丧失变体或部分功能丧失变体。因此,功能丧失变体是溶酶体基因或lsd相关联基因中的变体,其破坏溶酶体酶或其他整合的溶酶体蛋白的产生或正常功能。变体可以是缺失、取代、插入、剪接突变、启动子突变、导致稳定性改变的突变、移码或终止变体、无义突变和/或负面地影响溶酶体基因或其产生的蛋白质的正常功能的任何其他突变。例如,完全功能丧失、几乎完全功能丧失或部分功能丧失的变体可以导致酶的生产或活性的中断或降低。
65.完全功能丧失(lof)的变体可以被定义为预期与受影响的转录物的完全lof相关的一种或多种变体,诸如导致下游过早终止密码子或较大缺失的变体,该下游过早终止密码子或较大缺失去除受影响的转录物的第一外显子或超过50%的蛋白质编码序列(单倍体
不足性)(macarthur等人,2012《科学(science)》335,823
–
828)。几乎完全或部分lof变体会降低基因活性,但不会将其完全消融。
66.如本文所示,已发现许多包含与溶酶体贮积病(lsd)相关联的变体的基因与阿尔茨海默病(ad)和帕金森病(pd)密切相关。这是新的发现,因为受试者将需要对于这些突变是纯合的以便发展lsd,但受试者只需要是杂合的以便发展app加工的或微妙的溶酶体功能障碍相关的疾病,诸如ad或pd。
67.这一发现也很重要,因为先前没有认识到,对于这些基因而言,单倍体不足性且这些lsd蛋白的表达减少会与任何其他疾病状态相关联。如上所述,长期以来的观点认为,携带可以导致lsd的杂合突变的人(携带者)不容易患任何疾病。
68.如本文所示,溶酶体基因(例如,与lsd或正常alp功能相关联的基因)中的杂合有害突变与ad相关联。这些基因可以为ad发病机制中溶酶体功能障碍的机制提供更深入的了解,这是目前缺乏的知识并且该知识可以产生新的治疗靶点。所公开的结果可以为目前针对lsd的再利用治疗策略建立基础,以用于ad的潜在治疗。令人惊讶的是,nagu和ppt1模型(它们甚至不是在ad中鉴定出的具有有害变体的最显著富集基因)(参见,例如表13)显示对lsd疗法有应答。因此,向对其他经鉴定的在ad中显著富集的导致lsd的基因具有有害的杂合变体(或完全或几乎完全的功能丧失变体)的受试者提供的lsd治疗将被预期至少同样或更好地应答。
69.本文发现,在ad和pd患者中富集了45个具有完全或几乎完全功能丧失杂合变体的溶酶体酶基因。以下为已知的lsd相关联基因:aga、arsa、arsb、asah1、cln2(tpp1)、cln3、cln5、cln6、cln8、ctns、ctsa、ctsd、ctsk、fuca1、gaa、galc、galns、gla、glb1、gm2a、gnptab、gnptg、gns、gusb、hexa、hexb、hgsnat、hyal1、ids、idua、kctd7、lamp2、lipa、man2b1、manba、mcoln1、mfsd8、naga、naglu、neu1、npc1、npc2、ppt1、psap、sgsh、slc17a5、smpd1、sumf1、chit1、atp13a2、ctsf、dnajc5或grn。用于测量与这些基因相关联的酶缺陷和对这些基因的变体进行基因分型的测定在本领域中是公知的。
70.具有alp相关联基因中的缺陷的受试者的群体也可能具有溶酶体功能障碍。目前存在453个已知的溶酶体基因。因此,以下基因中的lof变体可能导致溶酶体功能障碍,并且可用如本文所述的疗法治疗:abca2、abca3、abca5、abcb9、abcc10、acp2、acp5、acpp、ada、adam8、adrb2、aga、ahnak、aldob、ankfy1、ankrd27、anpep、anxa11、ap1b1、ap1g1、ap1m1、ap1m2、ap1s1、ap1s2、ap1s3、ap3b1、ap3b2、ap3d1、ap3m1、ap3m2、ap3s1、ap3s2、ap4b1、ap4e1、ap4m1、ap4s1、aqp2、arf1、arl8a、arl8b、arrb1、arsa、arsb、arsd、arsg、asah1、ass1、atp11a、atp11c、atp13a2、atp6ap1、atp6v0a1、atp6v0a2、atp6v0a4、atp6v0b、atp6v0c、atp6v0d1、atp6v0d2、atp6v1a、atp6v1b1、atp6v1b2、atp6v1c1、atp6v1c2、atp6v1d、atp6v1e1、atp6v1f、atp6v1g1、atp6v1h、azu1、bcl10、bloc1s1、btd、c18orf8、c19orf28、c1orf85、c2orf18、c7orf28b、cat、ccdc115、cckar、ccz1、cd164、cd1b、cd1d、cd1e、cd63、cd68、cd74、cecr1、chid1、chit1、clcn5、clcn6、clcn7、cln3、cln5、clta、cltb、cltc、cltcl1、clu、col6a1、cp、cpvl、creg1、cst3、cst7、ctbs、ctns、ctsa、ctsb、ctsc、ctsd、ctse、ctsf、ctsg、ctsh、ctsk、ctsl1、ctsl2、ctso、ctss、ctsw、ctsz、cubn、cxcr2、cybasc3、daglb、depdc5、dkfzp761e198、dnajc13、dnajc5、dnajc6、dnase1、dnase2、dnase2b、dnm2、doc2a、dpp4、dpp7、dram1、dram2、ece1、egf、elane、enpep、enpp1、entpd4、epdr1、fam176a、fgfr3、
flot1、flot2、fnbp1、fuca1、fuca2、gaa、gabarap、galc、galns、gba、gc、gdap2、gga1、gga2、gga3、ggh、gja1、gla、glb1、gm2a、gna11、gnai1、gnai2、gnai3、gnaq、gnb1、gnb2、gnb4、gnptab、gnptg、gns、got1、gpc3、gpld1、gpr137、gpr137b、gpr143、grn、gusb、hexa、hexb、hgsnat、hla
‑
dma、hla
‑
dmb、hla
‑
doa、hla
‑
dob、hla
‑
dpa1、hla
‑
dpb1、hla
‑
dqa1、hla
‑
dqa2、hla
‑
dqb1、hla
‑
dqb2、hla
‑
dra、hla
‑
drb1、hla
‑
drb3、hla
‑
drb4、hla
‑
drb5、hps1、hps4、hpse、hspa8、hyal1、hyal2、hyal3、ids、idua、ifi30、igf2r、il4i1、itm2c、kcne1、kcne2、kiaa0226、kiaa0415、kiaa1609、lamp1、lamp2、lamp3、lamtor1、lamtor2、laptm4a、laptm4b、laptm5、ldlr、lgmn、lhcgr、lipa、litaf、lmbrd1、lnpep、loc653653、lrba、lrp1、lrp2、m6pr、man2b1、man2b2、manba、1
‑
mar、2
‑
mar、3
‑
mar、8
‑
mar、9
‑
mar、mcoln1、mcoln2、mcoln3、mfsd1、mfsd8、mios、mmd、mon1b、mpo、mtor、mylpf、myo7a、naaa、naga、naglu、nagpa、napa、napg、napsa、nbr1、ncstn、neu1、neu4、npc1、npc2、nppa、nsf、oca2、ostm1、p2rx4、p2ry2、pcsk9、pcyox1、pebp4、pgcp、pi4k2a、pla2g15、pla2g4e、pla2g4f、plbd1、plbd2、pld1、pld3、plekhf1、plod1、pnpla7、pon2、ppt1、ppt2、prcp、prdx6、prf1、prtn3、psap、psapl1、psen1、psen2、ptgds、rab14、rab27a、rab2a、rab5c、rab7a、rab7b、rab9a、ramp2、ramp3、rdh14、rilp、rnase1、rnase2、rnase6、rnaset2、rnf13、rnf152、rptor、rraga、rragb、rragc、rragd、scarb1、scarb2、scpep1、selrc1、serinc2、sftpb、sftpd、sgsh、sh3gl2、siae、sidt2、slc11a1、slc11a2、slc12a4、slc15a3、slc15a4、slc17a5、slc26a11、slc29a3、slc2a13、slc2a8、slc30a2、slc36a1、slc37a3、slc44a2、slc48a1、smcr8、smpd1、smpd4、smpdl3a、snap23、snx16、sort1、spaca3、spg11、sphk2、spns1、sppl2a、srgn、stard3、stard3nl、sts、stx3、stx7、stxbp2、sumf1、tcirg1、tial1、tlr3、tlr7、tlr9、tm9sf1、tmbim1、tmem127、tmem175、tmem192、tmem55a、tmem55b、tmem63a、tmem74、tmem8a、tmem9、tmem92、tmem97、tom1l1、tpcn1、tpcn2、tpp1、trim23、trip10、tspan1、tspan8、txndc5、tyr、uba52、unc13d、unc93b1、usp4、usp5、usp6、uvrag、vamp4、vamp7、vasn、vma21、vps11、vps16、vps18、vps33a、vps33b、vps35、vps36、vps39、vps41、vps4b、wdr11、wdr41、wdr48、zfyve26、znrf1或znrf2。
71.与自噬
‑
溶酶体途径(alp)功能障碍相关联的神经系统疾病、障碍和病症
72.本文发现,对于通常与溶酶体贮积病(lsd)相关联的完全的或几乎完全的功能丧失变体杂合的受试者(携带者)(当受试者对于这些基因变体是纯合的时)可以导致alp功能障碍或app加工缺陷。溶酶体基因中的有害变体可以引起亚临床溶酶体功能障碍,其可以导致改变的app加工(例如,改变的脑间质aβ水平和增加的aβ斑块负荷)或α
‑
syn聚集,特别是在患有阿尔茨海默病(ad)和帕金森病(pd)的受试者中。当将溶酶体基因(naglu、ppt1或csp
‑
α)中的杂合的完全或几乎完全的功能丧失的突变培育到ad或pd的小鼠模型上时,这是非常明显的。在这种情况下,杂合的完全或几乎完全的功能丧失的突变大大加剧了aβ斑块的形成或α
‑
突触核蛋白的聚集。因此,这里发现,亚临床溶酶体功能障碍可以导致神经系统疾病、障碍或病症。与溶酶体功能障碍相关联的其他神经系统疾病、障碍和病症可以是遗传性脑淀粉样血管病、以中风和智力功能下降为特征的病症(痴呆)、克雅氏病、运动神经元疾病、多聚谷氨酰胺障碍诸如亨廷顿病,以及外周组织疾病如家族性淀粉样多发性神经病(fap)、路易体痴呆、多系统萎缩或额颞叶痴呆。
73.本公开提供了用于治疗对于与溶酶体贮积病(lsd)相关联的基因变体杂合的受试者(例如,携带者)以及治疗或预防与自噬
‑
溶酶体途径(alp)相关联的神经系统或神经退行
性疾病、障碍或病症的方法。
74.例如,表13描述了具有在患有ad的受试者中的导致lsd的基因中发现的变体的基因。如果受试者对于这些基因是纯合的,则它们会发展lsd。但如本文中令人惊奇地发现的,杂合性在本文中与被诊断为患有自噬
‑
溶酶体途径相关联的神经系统或神经退行性疾病、障碍或病症(诸如ad或pd)的未来风险的预测相关联。
75.因此,所公开的诊断和治疗具有这些杂合的完全或几乎完全的功能丧失基因变体的受试者的方法可以用于检测或治疗与自噬
‑
溶酶体途径相关联的神经系统疾病状态。例如,该方法可以用于患有或被怀疑患有神经系统疾病、障碍或病症的受试者,该神经系统疾病、障碍或病症诸如与溶酶体或自噬功能障碍相关联的任何神经系统疾病或神经退行性疾病(例如,ad、pd、ftd等)。
76.对于具有完全或几乎完全的功能丧失变体的溶酶体贮积病相关联的或有害的溶酶体基因杂合的受试者中的其他神经系统或神经退行性疾病、障碍或病症可以通过本文所述的方法治疗。
77.溶酶体贮积病(lsd)
78.溶酶体贮积病(lsd)或障碍的特征是自噬
‑
溶酶体途径基因中的纯合的完全或几乎完全功能丧失的基因变体,导致溶酶体蛋白或溶酶体中的大分子的降解途径所必需的溶酶体蛋白功能的减少或完全丧失。如本文所述,发现杂合的完全或几乎完全功能丧失的溶酶体基因变体的受试者具有发生与app加工功能障碍(ad)或α
‑
突触核蛋白聚集(pd)相关联的神经系统疾病的风险。因此,可以实施对在新发现的神经系统溶酶体相关联疾病中由杂合性功能丧失变体引起的疾病的治疗。
79.lsd是一组至少50种遗传性疾病,其特征为参与溶酶体中大分子的降解途径的一种特异性溶酶体蛋白的全部或部分缺陷。它们是单基因的,并且对于它们中的大多数来说,已经描述了大量的突变。一些突变会导致蛋白功能的完全丧失,而其他突变只会降低正常功能。未降解的或部分降解的材料(通常是缺陷溶酶体酶的底物)的储存发生在溶酶体中。通常,lsd根据积累的非降解底物的化学性质进行分组,包括粘多糖病、脂沉积症、糖原病和寡糖症等。
80.尽管症状有很大的异质性,但这些疾病中的大多数疾病的特点是其病程进展,发作率高和死亡率增加,尽管在不同的疾病之间以及在患有同一疾病的患者之间存在显著差异。一般而言,这些疾病为多系统性的,并且临床特征包括器官肿大、中枢神经系统功能障碍以及毛发和面部粗糙。大多数患者在出生时无症状,并且在儿童期呈现发作。它们的频率在不同地区和人群中不同,但尽管个别罕见,但综合的估计的流行率在1:4000至1:9000活产之间。有趣的是,大多数儿童lsd具有显著的神经系统成分。
81.虽然这些疾病中的几种疾病到目前为止没有特定的疗法,但对于一些lsd,造血干细胞移植(hsct)、酶替代疗法(ert)、基因疗法(gt)和小分子药物是可用的或正在临床试验中。
82.自噬
‑
溶酶体途径功能增强剂:溶酶体贮积病治疗和疗法
83.本公开提供了对与lsd疾病、障碍或病症相关联的完全或几乎完全功能丧失溶酶体基因为杂合的受试者的鉴定和治疗。这些杂合的受试者处于具有或发展自噬
‑
溶酶体途径(alp)相关联的神经系统或神经退行性疾病、障碍或病症的较高的风险。本文已经显示本
lsd疗法和治疗通过挽救或增强自噬
‑
溶酶体途径(alp)功能,在动物模型中治疗或预防alp相关联的神经系统或神经退行性疾病、障碍或病症。
84.诸如基因疗法和酶替代疗法之类的lsd治疗已经显示在疾病的纯合子动物模型和人类患者中治疗lsd。因此,将预期与纯合子人群相比,这些疗法在杂合子人群中甚至更有效并且疾病更容易治疗。换言之,由于与杂合子人群(例如ad和pd)相关联的功能障碍与lsd患者相比较轻,因此与纯合子人群相比,在杂合子人群中治疗效果的阈值会较低。
85.治疗(lsd治疗剂)和用于lsd的治疗方法是众所周知的;参见,例如,ohashi 2018,“溶酶体贮积病和过氧化物酶体病的基因疗法(gene therapy for lysosomal storage diseases and peroxisomal diseases)”,《人类遗传学杂志(journal of human genetics)》(2019)64:139
‑
143;beck 2017“溶酶体贮积症的治疗策略(treatment strategies for lysosomal storage disorders)”,《发展医学和儿童神经病学(dev med&child neuro)》,13
‑
18;ferreira和gahl 2017,“溶酶体贮积病(lysosomal storage diseases)”,《罕见病转化科学(translational science of rare diseases)》2(1
‑
2)1
‑
71;platt 2017“清空仓库:溶酶体疾病和治疗策略(emptying the stores:lysosomal diseases and therapeutic strategies)”,《自然评论药物发现(nature reviews drug discovery)》17 133
‑
150;marques和saftig 2019,“溶酶体贮积症—挑战、概念和疗法途径:超越罕见病(lysosomal storage disorders
–
challenges,concepts and avenues for therapy:beyond rare diseases)”《细胞科学杂志(j cell sci)》132jcs221739。因此,除非本文另有说明,否则本公开的治疗和治疗方法可以根据此类过程进行。
86.表1.可以用作alp功能增强剂的溶酶体贮积病(lsd)的治疗策略。
87.[0088][0089]
表2.溶酶体贮积病(lsd)及相关联酶缺乏、治疗和基因。
[0090]
[0091]
[0092][0093]
因为已经发现用于lsd的这些和其他疗法对纯合子受试者有效,所以将预期它们在杂合子受试者中也会是有效的。
[0094]
底物减少疗法(srt)
[0095]
在代谢或遗传途径中,酶催化一系列反应。每种酶都通过其rna和蛋白质产物由基因进行调控或介导。在途径中的每个阶段,酶活性催化其中前体分子(底物)被转化为其下一个中间状态的反应。代谢途径的失败会导致底物的积累,并可能具有有害影响。底物减少疗法通过将底物的水平降低至其中残余降解活性足以防止底物积累的水平而解决了这种失败。
[0096]
底物减少疗法的基本原理是将溶酶体物质的形成减少到残余酶活性可以分解代谢储存和进入的溶酶体物质的速率。srt的示例可以包括美格鲁特(zavesca)或依利格鲁司他(cerdelga)。
[0097]
造血干细胞移植(hsct)
[0098]
在hsct中,来自健康供体的骨髓或脐带血的干细胞被移植。有证据表明,其疗效不仅依赖于供体细胞向骨髓中的迁移和血液谱系的重建,而且还依赖于移植细胞随后向许多疾病靶器官(包括脑)中的迁移,在那里它们取代了常驻的酶缺陷群体;从而成为功能性酶的局部和稳定来源。这通过通常被称为“交叉校正”的过程被进一步加强。交叉校正是这样一个过程,其中溶酶体酶可以从一个细胞(在这种情况下是供体造血细胞)分泌,并通过受体介导的过程被邻近细胞(造血或非造血来源的邻近细胞)吸收。在许多情况下,共享足够的酶以便完全校正与纯合子完全或几乎完全功能丧失突变相关联的生化缺陷。当成功时,hsct可以延长患者的生命,保留神经认知并且增强躯体改变。hsct的缺点包括与该程序相关联的重大风险,诸如发展移植物抗宿主病的可能性、难以找到hla兼容供体以及形成嵌合现象。因此,它在许多国家的使用已经被推迟,只要ert可获得,就使用ert。
[0099]
酶替代疗法(ert)
[0100]
在ert中,通过反复静脉注射的方式向患者施用缺陷重组酶。在这种情况下,重组酶通过“交叉校正”中涉及的相同受体介导的过程由细胞摄取。尽管ert是用于各种lsd的有效和安全的治疗选项,但ert也具有重要的局限性。其中包括由一些患者表现出的不良反应、高治疗成本、对每周4
‑
5小时输液的终身依赖以及校正神经系统和骨骼病理的有限的能力。
[0101]
基因疗法与基因组编辑
[0102]
基因疗法可以包括插入带有病毒载体的功能基因。溶酶体贮积病(lsd)的基因疗法进展迅速。大多数lsd的特征是脑受累(brain involvement),这促使靶向脑的疗法的开发。针对lsd中的脑受累,存在两种类型的基因疗法,即直接将治疗基因转移到脑细胞中和离体造血干细胞靶向的基因疗法。后一种方法的基本原理是脑小胶质细胞来源于造血细胞。因此,基因校正的造血细胞迁移到脑中并分化为小胶质细胞。这些经基因校正的小胶质细胞交叉校正与lsd相关联的代谢缺陷,并且减少了lsd中的炎症,产生临床益处。基因编辑技术也已被应用于该领域,并且目前正在进行聚焦于lsd的试验(参见,例如de carvalho等人,2015,“基因组编辑:溶酶体贮积病的潜在治疗(genome editing:potential treatment for lysosomal storage diseases)”,《当前干细胞报告(current stem cell reports)》1(1)9
‑
15)。尽管这些方法仍在调查中,但已经获得了非常令人鼓舞的结果。目前,在市场上存在几种经批准的基因疗法,包括用于lpl缺陷:aav/lpl(glyvera);ada缺乏:逆转录病毒/ada(strimvelis);和伯氏先天性黑内障(leber’s congenital amaurosis):aav/rpe65(luxturna)的基因疗法。
[0103]
最近,对于基因疗法存在改善的前景。例如,在2019年第一季度,有372项正在进行的基因疗法临床试验(再生医学联盟(alliance for regenerative medicine),5/9/19)。
[0104]
可以使用本领域中已知的任何载体。例如,该载体可以是选自逆转录病毒、慢病毒、疱疹病毒、腺病毒、腺相关病毒(aav)、狂犬病病毒、埃博拉病毒、慢病毒或其杂交体的病毒载体。
[0105]
表3.基因疗法策略。
[0106]
[0107][0108]
基因疗法可以允许酶直接持续递送至靶器官,并且消除对每周输注的需要。此外,少数细胞的校正可能导致酶被分泌到循环中并被其相邻细胞吸收(交叉校正),从而导致生化缺陷的广泛校正。因此,必须用基因转移载体修饰的细胞的数量相对较少。此外,精确的转录调节可能不是必需的,因为溶酶体酶的过表达似乎不是有害的,并且低至5
‑
10%的正常酶水平可以用于治疗几种lsd。
[0109]
遗传修饰可以离体或体内进行。离体策略是基于对培养物中的细胞进行修饰,并将修饰的细胞移植到患者体内。最常被认为是单基因疾病的治疗靶的细胞是干细胞。从各种来源收集和分离这些细胞的进展已经促进了自体基因疗法作为lsd的可行选项。在lsd的小鼠模型中,编码酶基因的遗传修饰的神经干细胞有效地减少了溶酶体储存,减少了病理,并且延长了动物的寿命。间充质干细胞和诱导型多能干细胞(ipsc)也用于此目的。然而,常规的基因疗法方案可能具有局限性;其中包括与免疫应答相关的安全性问题和在病毒载体
的情况下插入诱变的可能性,以及在非病毒载体的情况下的低效率。
[0110]
使用核酸内切酶进行靶向的基因组编辑可以解决常规基因疗法方案带来的局限性。这些酶是定制的分子剪刀,允许在几乎所有的细胞类型中,将dna切割成定义明确的、完全指定的片段。此外,它们可以通过瞬时表达核酸酶的质粒或通过转录的rna被递送至细胞,避免病毒的使用。
[0111]
联合疗法
[0112]
治疗方法的组合已经显示出对lsd有效。例如,本发明人先前已经表明,基因疗法、hst移植和小分子底物减少的组合在治疗克拉伯病中是最有效的。因此,对于患有溶酶体功能障碍相关联的神经系统疾病(诸如ad或pd)的受试者,类似的联合方法也预期是最有效的。但是,如本文先前所公开的,预期的是杂合群体将更容易治疗并且具有较低的治疗效力阈值,因为预期与杂合群体相关联的功能障碍远小于杂合lsd群体。
[0113]
作为另一个示例,基因疗法可以与hsct组合。从患者提取的造血干细胞可以用编码内切酶的载体和供体载体转染,该内切酶被设计成在接近特异性突变的位点裂解,供体载体含有与突变的区域同源的区域,然而具有正确的核苷酸序列,并且将作为双链断裂后dna损伤修复的模板。然后,内在化两个载体(其中发生切割和同源重组)的细胞将具有正确的基因序列,并且可以被选择和植入回患者体内。将自体hsct与核酸酶介导的基因组编辑相结合将具有降低患者治疗期间感染的风险的优势,因为免疫功能的恢复迅速。此外,由于供体和受体是同一个体,因此将避免排斥(移植物抗宿主病)的发展。校正的造血干细胞(hsc)治疗可以包括以下步骤:可以用编码定制内切酶的载体和供体载体转染从患者提取的造血干细胞,以指导同源重组。然后,可以离体选择校正的细胞并植入回患者体内。
[0114]
个性化的/精确药物
[0115]
溶酶体基因缺陷的杂合性导致改变的app加工和增加的aβ斑块沉积的发现允许个体化的治疗方法。检测溶酶体相关联基因中完全或几乎完全功能丧失变体的方法可以用作基于该信息治疗它们的基础(参见,例如表1、表2和表3)。
[0116]
现在,可以根据检测到的溶酶体基因变体来鉴定和治疗有与溶酶体功能障碍相关联的神经系统疾病风险的受试者。溶酶体基因缺陷和lsd的治疗是众所周知的,并且已显示出有效地缓解与溶酶体功能障碍相关联的神经系统疾病(例如ad)的影响。
[0117]
连同本文提供的证据,目前用于患有lsd的纯合子儿童的lsd的治疗预期将在有风险的携带者(杂合子)中发挥作用,因为两者都涉及自噬
‑
溶酶体途径,并且对于杂合子受试者,疗效的阈值可能低得多。
[0118]
自噬
‑
溶酶体途径(alp)
[0119]
本文描述了用于调节自噬
‑
溶酶体途径(alp)以用于治疗神经系统或神经退行性疾病、障碍和病症的方法。
[0120]
自噬
‑
溶酶体途径(alp)是细胞内细胞器和易聚集蛋白降解的主要途径。自噬(“自我吞噬”)是一种细胞内降解途径,其负责经由溶酶体消化和回收营养物质。越来越多的证据表明,溶酶体功能障碍可以在多种神经退行性疾病中起作用,最显著的是阿尔茨海默病(ad)和帕金森病(pd)。溶酶体基因功能丧失变体也可能与病因不明的痴呆病例相关联。
[0121]
如本文所述,在人类基因组中存在至少430个与alp相关联的基因(38个自噬基因、161个自噬调节基因、64个溶酶体基因和167个溶酶体调节基因)。自噬
‑
溶酶体途径相关联
基因中携带杂合子有害变体的个体有发展常见成人发作神经系统疾病(例如阿尔茨海默病、帕金森病、额颞叶痴呆等)的风险。此外,通过酶替代疗法(ert)、基因疗法(gt)、干细胞疗法等补充外源性溶酶体蛋白可以减缓这些疾病的进展。
[0122]
如本文所述,通过鉴定pd和ad中alp基因的特异性缺陷,可以利用包括基因疗法、酶替代、口服小分子底物减少疗法、小分子伴侣和自噬途径的药理学补救的多种治疗策略以用于pd和ad以及其他神经和神经退行性疾病和病症的潜在治疗。如本文所述,可以调节与alp相关联的基因的表达以用于治疗神经系统或神经退行性疾病或障碍。来自与alp相关联的基因的蛋白产物也可以通过酶替代疗法(ert)进行补充。
[0123]
与lsd相关联的基因可以包括但不限于:aga、arsa、arsb、asah1、cln2(tpp1)、cln3、cln5、cln6、cln8、ctns、ctsa、ctsd、ctsk、fuca1、gaa、galc、galns、gla、glb1、gm2a、gnptab、gnptg、gns、gusb、hexa、hexb、hgsnat、hyal1、ids、idua、kctd7、lamp2、lipa、man2b1、manba、mcoln1、mfsd8、naga、naglu、neu1、npc1、npc2、ppt1、psap、sgsh、slc17a5、smpd1、sumf1、chit1、atp13a2、ctsf、dnajc5或grn。例如,与成人发作的神经退行性疾病相关的lsd相关联基因可以是但不限于:asah1、cln3、cln8、cstd、ctns、ctsa ctsf、dnajc5、gaa、galc、galns、gba、gla、glb1、gnptab、gns、grn、hexb、hgsnat、ids、idua、man2b1、manba、mfsd8、naglu、neu1、npc1、npc2、pld3、ppt1、sgsh、smpd1、sorl1和tpp1。
[0124]
与alp相关联的基因包括但不限于:abca2、abca3、abca5、abcb9、abcc10、acp2、acp5、acpp、ada、adam8、adrb2、aga、ahnak、aldob、ankfy1、ankrd27、anpep、anxa11、ap1b1、ap1g1、ap1m1、ap1m2、ap1s1、ap1s2、ap1s3、ap3b1、ap3b2、ap3d1、ap3m1、ap3m2、ap3s1、ap3s2、ap4b1、ap4e1、ap4m1、ap4s1、aqp2、arf1、arl8a、arl8b、arrb1、arsa、arsb、arsd、arsg、asah1、ass1、atp11a、atp11c、atp13a2、atp6ap1、atp6v0a1、atp6v0a2、atp6v0a4、atp6v0b、atp6v0c、atp6v0d1、atp6v0d2、atp6v1a、atp6v1b1、atp6v1b2、atp6v1c1、atp6v1c2、atp6v1d、atp6v1e1、atp6v1f、atp6v1g1、atp6v1h、azu1、bcl10、bloc1s1、btd、c18orf8、c19orf28、c1orf85、c2orf18、c7orf28b、cat、ccdc115、cckar、ccz1、cd164、cd1b、cd1d、cd1e、cd63、cd68、cd74、cecr1、chid1、chit1、clcn5、clcn6、clcn7、cln3、cln5、clta、cltb、cltc、cltcl1、clu、col6a1、cp、cpvl、creg1、cst3、cst7、ctbs、ctns、ctsa、ctsb、ctsc、ctsd、ctse、ctsf、ctsg、ctsh、ctsk、ctsl1、ctsl2、ctso、ctss、ctsw、ctsz、cubn、cxcr2、cybasc3、daglb、depdc5、dkfzp761e198、dnajc13、dnajc5、dnajc6、dnase1、dnase2、dnase2b、dnm2、doc2a、dpp4、dpp7、dram1、dram2、ece1、egf、elane、enpep、enpp1、entpd4、epdr1、fam176a、fgfr3、flot1、flot2、fnbp1、fuca1、fuca2、gaa、gabarap、galc、galns、gba、gc、gdap2、gga1、gga2、gga3、ggh、gja1、gla、glb1、gm2a、gna11、gnai1、gnai2、gnai3、gnaq、gnb1、gnb2、gnb4、gnptab、gnptg、gns、got1、gpc3、gpld1、gpr137、gpr137b、gpr143、grn、gusb、hexa、hexb、hgsnat、hla
‑
dma、hla
‑
dmb、hla
‑
doa、hla
‑
dob、hla
‑
dpa1、hla
‑
dpb1、hla
‑
dqa1、hla
‑
dqa2、hla
‑
dqb1、hla
‑
dqb2、hla
‑
dra、hla
‑
drb1、hla
‑
drb3、hla
‑
drb4、hla
‑
drb5、hps1、hps4、hpse、hspa8、hyal1、hyal2、hyal3、ids、idua、ifi30、igf2r、il4i1、itm2c、kcne1、kcne2、kiaa0226、kiaa0415、kiaa1609、lamp1、lamp2、lamp3、lamtor1、lamtor2、laptm4a、laptm4b、laptm5、ldlr、lgmn、lhcgr、lipa、litaf、lmbrd1、lnpep、loc653653、lrba、lrp1、lrp2、m6pr、man2b1、man2b2、manba、1
‑
mar、2
‑
mar、3
‑
mar、8
‑
mar、9
‑
mar、mcoln1、mcoln2、mcoln3、mfsd1、mfsd8、mios、mmd、mon1b、mpo、mtor、mylpf、myo7a、naaa、naga、
naglu、nagpa、napa、napg、napsa、nbr1、ncstn、neu1、neu4、npc1、npc2、nppa、nsf、oca2、ostm1、p2rx4、p2ry2、pcsk9、pcyox1、pebp4、pgcp、pi4k2a、pla2g15、pla2g4e、pla2g4f、plbd1、plbd2、pld1、pld3、plekhf1、plod1、pnpla7、pon2、ppt1、ppt2、prcp、prdx6、prf1、prtn3、psap、psapl1、psen1、psen2、ptgds、rab14、rab27a、rab2a、rab5c、rab7a、rab7b、rab9a、ramp2、ramp3、rdh14、rilp、rnase1、rnase2、rnase6、rnaset2、rnf13、rnf152、rptor、rraga、rragb、rragc、rragd、scarb1、scarb2、scpep1、selrc1、serinc2、sftpb、sftpd、sgsh、sh3gl2、siae、sidt2、slc11a1、slc11a2、slc12a4、slc15a3、slc15a4、slc17a5、slc26a11、slc29a3、slc2a13、slc2a8、slc30a2、slc36a1、slc37a3、slc44a2、slc48a1、smcr8、smpd1、smpd4、smpdl3a、snap23、snx16、sort1、spaca3、spg11、sphk2、spns1、sppl2a、srgn、stard3、stard3nl、sts、stx3、stx7、stxbp2、sumf1、tcirg1、tial1、tlr3、tlr7、tlr9、tm9sf1、tmbim1、tmem127、tmem175、tmem192、tmem55a、tmem55b、tmem63a、tmem74、tmem8a、tmem9、tmem92、tmem97、tom1l1、tpcn1、tpcn2、tpp1、trim23、trip10、tspan1、tspan8、txndc5、tyr、uba52、unc13d、unc93b1、usp4、usp5、usp6、uvrag、vamp4、vamp7、vasn、vma21、vps11、vps16、vps18、vps33a、vps33b、vps35、vps36、vps39、vps41、vps4b、wdr11、wdr41、wdr48、zfyve26、znrf1或znrf2。
[0125]
分子工程改造
[0126]
提供以下定义和方法是为了更好地定义本发明并指导本领域普通技术人员实施本发明。除非另有说明,否则术语应由相关领域的普通技术人员根据常规用法来理解。
[0127]
如本文所用的术语“异源dna序列”、“外源性dna片段”或“异源核酸”均指来源于对特定宿主细胞是外源性的来源的序列,或者,如果来源于同一来源,则是从其原始形式修饰的序列。因此,宿主细胞中的异源基因包括对特定宿主细胞是内源性的但已经通过例如使用dna改组被修饰的基因。该术语还包括天然存在的dna序列的非天然存在的多个拷贝。因此,该术语指的是对细胞是外源性的或异源的、或与细胞同源但位于宿主细胞核酸中通常未发现该元件的位置的dna片段。外源性dna片段被表达以产生外源性多肽。“同源”dna序列是与导入该序列的宿主细胞天然相关联的dna序列。
[0128]
表达载体、表达构建体、质粒或重组dna构建体通常被理解为是指经由人类干预(包括通过重组手段或直接化学合成)产生的核酸,其具有一系列允许特定核酸在例如宿主细胞中转录或翻译的特定核酸元件。表达载体可以是质粒、病毒或核酸片段的一部分。通常,表达载体可以包括与启动子可操作连接的待转录的核酸。
[0129]“启动子”通常被理解为指导核酸的转录的核酸控制序列。诱导型启动子通常被理解为响应于特定刺激而介导可操作连接的基因的转录的启动子。启动子可以包括转录的起始位点附近的必需核酸序列,诸如,在聚合酶ii型启动子的情况下,tata元件。启动子可以任选地包括末端增强子或阻遏子元件,其可以位于距转录的起始位点多达几千个碱基对的位置。
[0130]
如本文所使用的“可转录核酸分子”是指能够被转录成rna分子的任何核酸分子。已知用于以可转录核酸分子被转录成功能性mrna分子的方式将构建体引入细胞的方法,该功能性mrna分子被翻译并且因此被表达为蛋白质产物。也可以构建能够表达反义rna分子的构建体,以便抑制感兴趣的特定rna分子的翻译。为了实施本公开,用于制备和使用构建体和宿主细胞的常规组合物和方法是本领域技术人员熟知的(参见,例如sambrook和
russel(2006),“来自分子克隆的浓缩方案:实验室手册(condensed protocols from molecular cloning:a laboratory manual)”,冷泉港实验室出版社(cold spring harbor laboratory press),isbn
‑
10:0879697717;ausubel等人(2002)《分子生物学中的短协议(short protocols in molecular biology)》,第5版,《当前协议(current protocols)》,isbn
‑
10:0471250929;sambrook和russel(2001)《分子克隆:实验室手册(molecular cloning:a laboratory manual)》,第3版,冷泉港实验室出版社,isbn
‑
10:0879695773;elhai,j.和wolk,c.p.1988.《酶学方法(methods in enzymology)》167,747
‑
754)。
[0131]“转录起始位点”或“起始位点”是作为转录的序列的一部分的第一个核苷酸周围的位置,其也被定义为位置+1。对于该位点,该基因及其控制区的所有其他序列都可以被编号。下游序列(即,在3’方向上的另外的蛋白质编码序列)可以被命名为阳性,而上游序列(在5’方向上的大多数控制区)被命名为阴性。
[0132]“可操作地连接的”或“功能性连接的”优选地是指核酸序列在单个核酸片段上的缔合,使得一个的功能受到另一个的影响。例如,如果调节性dna序列与编码rna或多肽的dna序列的位置使得调节性dna序列影响编码dna序列的表达(即编码序列或功能性rna在启动子的转录控制之下),则该调节性dna序列被称为与该dna序列“可操作地连接”或“缔合”。编码序列可以以有义或反义方向与调节性序列可操作地连接。两个核酸分子可以是单个连续核酸分子的一部分,并且可以是相邻的。例如,如果启动子调控或介导细胞中目的基因的转录,则启动子与目的基因可操作地连接。
[0133]“构建体”通常被理解为任何重组核酸分子,诸如质粒、粘粒、病毒、自主复制核酸分子、噬菌体、或线性或环状单链或双链dna或rna核酸分子,其来源于任何来源,能够基因组整合或自主复制,包含核酸分子,其中一个或多个核酸分子已被可操作地连接。
[0134]
本公开的构建体可以含有与可转录核酸分子可操作地连接的启动子,该可转录核酸分子与3’转录终止核酸分子可操作地连接。此外,构建体可以包括但不限于来自例如3
’‑
非翻译区(3’utr)的另外的调节性核酸分子。构建体可以包括但不限于mrna核酸分子的5’非翻译区(5’utr),其可以在翻译起始中起重要作用,并且也可以是表达构建体中的遗传组分。这些额外的上游和下游调节核酸分子可以来源于相对于启动子构建体上存在的其他元件是天然的或异源的来源。
[0135]
术语“转化”是指将核酸片段转移到宿主细胞的基因组中,产生遗传稳定的遗传。含有转化的核酸片段的宿主细胞被称为“转基因”细胞,并且包含转基因细胞的生物体被称为“转基因生物体”。
[0136]“转化的”、“转基因的”和“重组的”是指已经引入异源核酸分子的宿主细胞或生物体,诸如细菌、蓝细菌、动物或植物。核酸分子可以被稳定地整合到基因组中,如本领域通常已知和公开的(sambrook 1989;innis 1995;gelfand 1995;innis&gelfand 1999)。已知的pcr方法包括但不限于使用配对引物、巢式引物、单特异性引物、简并引物、基因特异性引物、载体特异性引物、部分错配引物等的方法。术语“未转化的”是指未经过转化过程的正常细胞。
[0137]“野生型”是指在自然界中发现的没有任何已知突变的病毒或生物体。
[0138]
具有上述所需的百分比同一性并保持所表达的蛋白的所需活性的变体核苷酸及其编码的多肽的设计、产生和测试在本领域的技术范围内。例如,突变体的定向进化和快速
分离可以根据参考文献中描述的方法进行,该参考文献包括但不限于link等人(2007)《自然评论(nature reviews)》5(9),680
‑
688;sanger等人(1991)《基因(gene)》97(1),119
‑
123;ghadessy等人(2001)《美国科学院院刊(proc natl acad sci usa)》98(8)4552
‑
4557。因此,本领域技术人员可以生产大量与本文所述的参考序列具有例如至少95
‑
99%同一性的核苷酸和/或多肽变体,并且根据本领域的常规方法筛选所需的表型。
[0139]
核苷酸和/或氨基酸序列同一性百分比(%)被理解为当两个序列比对时,与候选序列中的核苷酸或氨基酸残基相同的核苷酸或氨基酸残基与参考序列相比的百分比。为了确定同一性百分比,对序列进行比对,并且如有必要,引入间隙以获得最大的序列同一性百分比。确定同一性百分比的序列比对程序是本领域技术人员熟知的。通常使用公共可用的计算机软件,诸如blast、blast2、align2或megalign(dnastar)软件对序列进行比对。本领域技术人员可以确定用于测量比对的适当参数,包括在被比较的序列的全长上实现最大比对所需的任何算法。当对序列进行比对时,给定的序列a对给定的序列b、给定的序列a与给定的序列b、给定的序列a相对于给定的序列b的序列同一性百分比(其可以可替代地表述为给定的序列a对给定的序列b、与给定的序列b或相对于给定的序列b具有一定的序列同一性百分比,或者给定的序列a包括对给定的序列b、与给定的序列b或相对于给定的序列b的一定的序列同一性百分比)可以被计算为:序列同一性百分比=x/y100,其中x是通过序列比对程序或算法对a和b的比对被评为完全匹配的残基的数量,并且y是b中的残基的总数。如果序列a的长度不等于序列b的长度,则a对b的序列同一性百分比将不等于b对a的序列同一性百分比。
[0140]
一般而言,只要保留所需的活性,可以在任何位置进行保守取代。可以进行所谓的保守交换,其中被替换的氨基酸具有与原始氨基酸相似的性质,例如通过asp交换glu,通过asn交换gln,通过ile交换val,通过ile交换leu,以及通过thr交换ser。例如,具有相似性质的氨基酸可以是脂族氨基酸(例如,甘氨酸、丙氨酸、缬氨酸、亮氨酸、异亮氨酸);含羟基或硫/硒的氨基酸(例如,丝氨酸、半胱氨酸、硒代半胱氨酸、苏氨酸、蛋氨酸);环状氨基酸(例如,脯氨酸);芳族氨基酸(例如,苯丙氨酸、酪氨酸、色氨酸);碱性氨基酸(例如,组氨酸、赖氨酸、精氨酸);或酸性及其酰胺(例如,天冬氨酸、谷氨酸、天冬酰胺、谷氨酰胺)。缺失是氨基酸被直接键取代。缺失的位置包括多肽的末端和单独的蛋白结构域之间的连接。插入是将氨基酸引入到多肽链中,一个直接键被一个或多个氨基酸正式取代。可以借助于本领域已知的计算机模拟程序来调节氨基酸序列,该计算机模拟程序可以产生具有例如改进的活性或改变的调节的多肽。在这种人工产生的多肽序列的基础上,可以使用所需宿主细胞的特定密码子使用在体外合成编码此类调节的多肽的相应核酸分子。
[0141]“高度严格的杂交条件”被定义为在65℃下在6
×
ssc缓冲液(即0.9m的氯化钠和0.09m的柠檬酸钠)中进行的杂交。在这些条件下,可以通过计算两个序列之间的dna双链体的解链温度(t
m
)来确定给定的一组序列是否将杂交。如果在6
×
ssc的盐条件下,特定的双链体具有低于65℃的解链温度,则这两个序列将不会杂交。另一方面,如果在相同的盐条件下,解链温度高于65℃,则序列将杂交。一般而言,任何杂交的dna:dna序列的解链温度可以使用以下公式来确定:t
m
=81.5℃+16.6(log
10
[na
+
])+0.41(级分g/c含量)
–
0.63(%甲酰胺)
–
(600/l)。此外,每降低1%核苷酸同一性,dna:dna杂交体的t
m
被降低1
‑
1.5℃(参见,例如sambrook和russel,2006)。
[0142]
可以使用本领域已知的多种标准技术来转化宿主细胞(参见,例如sambrook和russel(2006),“来自分子克隆的浓缩方案:实验室手册”,冷泉港实验室出版社,isbn
‑
10:0879697717;ausubel等人.(2002)《分子生物学中的短协议》,第5版,《当前协议》,isbn
‑
10:0471250929;sambrook和russel(2001),《分子克隆:实验室手册》,第3版,冷泉港实验室出版社,isbn
‑
10:0879695773;elhai,j.和wolk,c.p.1988.《酶学方法》167,747
‑
754)。此类技术包括但不限于病毒感染、磷酸钙转染、脂质体介导的转染、微弹介导的递送、受体介导的摄取、细胞融合、电穿孔等。可以选择和增殖转染的细胞以提供重组宿主细胞,该重组宿主细胞包含被稳定地整合在宿主细胞基因组中的表达载体。
[0143][0144][0145]
[0146][0147]
可以引入到宿主细胞中的示例性核酸包括,例如,来自另一物种的dna序列或基因,或甚至源自或存在于相同物种中但通过基因工程改造方法并入受体细胞中的基因或序列。术语“外源性的”也旨在是指通常不存在于被转化的细胞中,或者可能只是不以如在转化的dna片段或基因中发现的形式、结构等存在的基因,或正常存在并希望以不同于天然表达模式的方式表达(例如过表达)的基因。因此,术语“外源性”基因或dna旨在是指被引入到受体细胞中的任何基因或dna片段,而不管在此类细胞中是否可能已经存在类似的基因。外源性dna中包括的dna的类型可以包括在细胞中已经存在的dna、来自相同类型生物体的另一个个体的dna、来自不同生物体的dna、或外部产生的dna,诸如含有基因的反义消息的dna序列、或编码基因的合成或修饰版本的dna序列。
[0148]
根据本文所述的方法开发的宿主菌株可以通过本领域已知的多种方法进行评价(参见,例如,studier(2005)《蛋白质的表达和纯化(protein expr purif.)》41(1),207
–
234;gellissen编辑.(2005)《重组蛋白的生产:新型微生物和真核表达系统(production of recombinant proteins:novel microbial and eukaryotic expression systems)》,wiley
‑
vch,isbn
‑
10:3527310363;baneyx(2004)《蛋白质表达技术(protein expression technologies)》,taylor&francis,isbn
‑
10:0954523253)。
[0149]
下调或沉默基因的方法在本领域中是已知的。例如,表达的蛋白质活性可以使用反义寡核苷酸、蛋白质适体、核苷酸适体和rna干扰(rnai)(例如,小干扰rna(sirna)、短发夹rna(shrna)和微rna(mirna)下调或消除(参见,例如,fanning和symonds(2006)《实验药理学手册(handb exp pharmacol.)》173,289
‑
303g,其描述锤头状核酶和小发夹rna;helene,c.等人.(1992)《纽约科学院年鉴(ann.n.y.acad.sci.)》660,27
‑
36;maher(1992)《生物测定(bioassays)》14(12):807
‑
15,其描述靶向脱氧核糖核苷酸序列;lee等人.(2006)《化学生物学的最新观点(curr opin chem biol.)》10,1
‑
8,其描述了适体;
reynolds等人.(2004)《自然生物技术(nature biotechnology)》22(3),326
–
330,其描述了rnai;pushparaj和melendez(2006)《临床和实验药理学和生理学(clinical and experimental pharmacology and physiology)》33(5
‑
6),504
‑
510,其描述了rnai;dillon等人.(2005)《生理学年度评论(annual review of physiology)》67,147
‑
173,其描述了rnai;dykxhoorn和lieberman(2005)《医学年度评论(annual review of medicine)》56,401
‑
423,其描述了rnai)。rnai分子可以从各种来源商购(例如,ambion,tx;西格玛奥德里奇(sigma aldrich),mo;英杰公司(invitrogen)。使用各种算法的几个sirna分子设计程序是本领域已知的(参见,例如cenix算法,ambion;block
‑
it
tm rnai设计者,英杰公司;sirna
·
怀特黑德研究所设计工具,生物数学与研究计算(sirna whitehead institute design tools,bioinofrmatics&research computing))。对确定最佳sirna序列有影响的性状包括sirna的末端的g/c含量、sirna的特定内部结构域的tm、sirna长度、cds(编码区)内靶序列的位置以及3’突出端的核苷酸含量。
[0150]
基因组编辑
[0151]
使用工程改造的锌指核酸酶(zfn)、转录激活子样效应子核酸酶(talen)以及最近成簇的规律间隔的短回文重复序列
‑
crispr相关蛋白9(crispr
‑
cas9)系统的基因组编辑技术的最新进展已经使得能实现精确修饰基因组中的靶位点的可能性。这项技术带来了治愈许多遗传性疾病的希望。这里,靶向的基因组编辑可以用于或与造血干细胞移植和其他方法结合用于治疗对功能性溶酶体基因变体的缺失杂合的受试者。
[0152]
如本文所述,可以使用基因组编辑来增强或增加酶活性。用于基因组编辑的工艺是众所周知的;参见,例如aldi 2018《自然通讯(nature communications)》9(1911)。因此,除非本文另有说明,否则本公开的工艺可以根据此类工艺进行。
[0153]
例如,基因组编辑可以包含crispr/cas9、crispr
‑
cpf1、talen或znf。通过基因组编辑对酶活性的充分增强可以导致保护而不受经受lsd。
[0154]
作为一个示例,成簇的规律间隔的短回文重复序列(crispr)/crispr相关(cas)系统是一类针对哺乳动物细胞中所需的基因组位点的新型基因组编辑工具。最近发表的ii型crispr/cas系统使用cas9核酸酶,该cas9核酸酶通过与合成导向rna络合而靶向基因组位点,该合成导向rna与20核苷酸dna序列杂交,并紧接在由cas9识别的ngg基序(因此为(n)
20
ngg靶dna序列)之前。这导致ngg基序上游三个核苷酸的双链断裂。双链断裂引发非同源的末端连接(这是容易出错的,并且有利于敲除基因等位基因的移码突变)或同源定向修复(这可以通过使用外源性引入的双链或单链dna修复模板被利用以敲入或校正基因组中的突变)。因此,基因组编辑(例如,使用crispr/cas系统)可能是用于治疗应用的有用工具,以用于治疗对于因酶产生或活性的增强或增加的功能性溶酶体基因变体缺失杂合的受试者。
[0155]
例如,如本文所述的方法可以包含用于改变细胞中靶多核苷酸序列的方法,该方法包含使多核苷酸序列与成簇的规律间隔的短回文重复序列相关(cas)蛋白接触。
[0156]
制剂
[0157]
本文所述的药剂和组合物可以使用一种或多种药学上可接受的载体或赋形剂通过任何常规的方式配制,如在例如雷明顿药物科学(remington’s pharmaceutical sciences)(a.r.gennaro编辑),第21版,isbn:0781746736(2005)(其通过引用以其整体并入本文)中所描述的。此类制剂将含有治疗有效量的本文所述的生物活性药剂(其可以是纯
化的形式),以及合适量的载体,以便提供用于向受试者适当施用的形式。
[0158]
术语“制剂”是指以适于施用于受试者诸如人的形式制备药物。因此,“制剂”可以包括药学上可接受的赋形剂,包括稀释剂或载体,诸如衣壳蛋白。
[0159]
如本文使用的术语“药学上可接受的”可以描述不会引起不可接受的药理学活性损失或不可接受的不利副作用的物质或组分。药学上可接受的成分的示例可以是在美国药典(united states pharmacopeia)(usp 29)和国家处方集(national formulary)(nf 24)、美国药典惯例公司(united states pharmacopeial convention,inc),马里兰州洛克维尔,2005(“usp/nf”)或更新版本中有专著的成分,以及在fda的持续更新的非活性成分搜索在线数据库中列出的组分。也可以使用usp/nf等中未描述的其他有用组分。
[0160]
如本文使用的术语“药学上可接受的赋形剂”可以包括任何和所有的溶剂、分散介质、包衣、抗细菌剂和抗真菌剂、等渗剂或吸收延迟剂。使用此类介质和药剂用于药物活性物质在本领域中是熟知的(通常参见雷明顿药物科学(a.r.gennaro编辑),第21版,isbn:0781746736(2005))。除了任何常规介质或药剂与活性成分不相容之外,其在治疗组合物中的应用也被预期。补充活性成分也可以被并入组合物中。
[0161]“稳定的”制剂或组合物可以指具有足够稳定性的组合物,以允许在方便的温度(诸如在约0℃与约60℃之间)下储存持续商业上合理的时间段,诸如至少约一天、至少约一周、至少约一个月、至少约三个月、至少约六个月、至少约一年或至少约两年。
[0162]
制剂应当适合施用方式。用于本公开的药剂可以通过已知的方法配制,用于使用几种途径对受试者施用,该途径包括但不限于鞘内(例如,基因疗法)、颅内(例如,基因疗法)、肠胃外、肺、口服、局部、皮内、瘤内、鼻内、吸入(例如,在气雾剂中)、植入、肌内、腹膜内、静脉内(例如,酶替代疗法)、脑室内、皮下、鼻内、硬膜外、眼内、经皮、颊和直肠。单独的药剂也可以与一种或多种另外的药剂联合施用,或与其他生物活性药剂或生物惰性药剂一起施用。此类生物活性药剂或生物惰性药剂可以与药剂流体或机制连通,或通过离子力、共价力、范德华力、疏水力、亲水力或其他物理力与药剂连接。
[0163]
可以配制控释(或缓释)制剂,以延长药剂的活性并降低给药频率。控释制剂也可以用于影响作用的开始时间或其他特征,诸如药剂的血液水平,并且从而影响副作用的发生。控释制剂可以被设计为最初释放一定量的产生所需治疗效果的药剂,并且逐渐和持续地释放其他量的药剂,以在延长的时间段内维持治疗效果的水平。为了维持药剂在体内接近恒定的水平,药剂可以从剂型中以一定的速率被释放,该速率将取代由机体代谢或排泄的药剂的量。药剂的控释可以由各种诱导剂刺激,例如ph的变化、温度的变化、酶、水或其他生理条件或分子。
[0164]
本文所述的药剂或组合物也可以与如下文进一步描述的其他治疗形式组合使用。因此,除了本文所述的疗法之外,还可以向受试者提供已知对疾病、障碍或病症的治疗有效的其他疗法。
[0165]
治疗方法
[0166]
还提供了一种治疗、预防或逆转在对与溶酶体功能障碍相关联的功能丧失基因变体杂合的受试者中的神经系统或神经退行性疾病、障碍或病症的方法,该受试者患有与增加的aβ或app加工功能障碍相关联的神经系统疾病、障碍或病症,疑似患有神经系统疾病、障碍或病症,或者有发展神经系统疾病、障碍或病症的风险,其中在需要的受试者中施用治
疗有效量的alp功能增强剂(例如,lsd治疗剂),以便基本上抑制神经系统疾病、障碍或病症,减缓神经系统疾病、障碍或病症的进展,或限制神经系统疾病、障碍或病症的发展。
[0167]
本文所述的方法通常对需要其的受试者进行。需要本文所述的治疗方法的受试者可以是患有、被诊断患有、疑似患有神经系统疾病、障碍或病症,或者有发展神经系统疾病、障碍或病症的风险的受试者。通常将通过与所讨论的疾病或病症相符的病史和体格检查来评估对治疗需要的确定。可通过本文所述的方法治疗的各种病症的诊断在本领域技术范围内。受试者可以是动物受试者,包括哺乳动物,诸如马、牛、狗、猫、羊、猪、小鼠、大鼠、猴、仓鼠、豚鼠和人。例如,受试者可以是人类受试者。
[0168]
一般而言,alp功能增强剂的安全且有效的量是例如在受试者中会引起期望的治疗效果同时使不期望的副作用最小化的量。在各种实施例中,本文所述的有效量的alp功能增强剂可以基本上抑制神经系统疾病、障碍或病症,减缓神经系统疾病、障碍或病症的进展,或限制神经系统疾病、障碍或病症的发展。
[0169]
根据本文所述的方法,施用可以是肠胃外、肺部、口服、局部、皮内、肌内、腹膜内、静脉内、鞘内、颅内、脑室内、皮下、鼻内、硬膜外、眼、颊或直肠施用。
[0170]
当用于本文所述的治疗中时,治疗有效量的alp功能增强剂可以以纯形式使用,或者如果存在此类形式,可以以药学上可接受的盐形式使用,并且具有或没有药学上可接受的赋形剂。例如,本公开的化合物可以以适用于任何医学治疗的合理受益/风险比以足以基本上抑制神经系统疾病、障碍或病症,减慢神经系统疾病、障碍或病症的进展或限制神经系统疾病、障碍或病症的发展的量施用。
[0171]
可以与药学上可接受的载体组合以产生单一剂型的本文所述的组合物的量将根据所治疗的宿主和特定的施用模式而变化。本领域技术人员将理解,含在每种剂型的单独剂量中的药剂的单位含量不需要本身构成治疗有效量,因为通过施用许多单独的剂量可以达到必要的治疗有效量。
[0172]
本文所述的组合物的毒性和治疗功效可以通过在细胞培养物或实验动物中的标准制药程序来确定,以用于确定ld
50
(对50%的群体致死的剂量)和ed
50
(对50%的群体治疗有效的剂量)。毒性与治疗效果之间的剂量比是可以被表示为比率ld
50
/ed
50
的治疗指数,其中较大的治疗指数在本领域中通常被理解为是最佳的。
[0173]
任何特定受试者的特定治疗有效剂量水平将取决于多种因素,包括所治疗的障碍和障碍的严重程度;所用的特定化合物的活性;所用的具体组合物;受试者的年龄、体重、一般健康状况、性别和饮食;施用时间;施用途径;所用的组合物的排泄速率;治疗的持续时间;与所用的特定化合物组合或巧合使用的药物;和医学领域中熟知的类似因素(参见,例如,koda
‑
kimble等人(2004)《应用治疗学:药物的临床应用(applied therapeutics:the clinical use of drugs)》,lippincott williams&wilkins,isbn0781748453;winter(2003)《基本临床药代动力学(basic clinical pharmacokinetics)》第4版,lippincott williams&wilkins,isbn 0781741475;sharqel(2004)《应用生物药剂学与药代动力学(applied biopharmaceutics&pharmacokinetics)》,mcgraw
‑
hill/appleton&lange,isbn 0071375503)。例如,以低于达到所需治疗效果所需水平的水平开始组合物的剂量并逐渐增加剂量直到达到所需的效果是本领域技术所熟知的。如果需要,出于施用目的,可以将有效日剂量分成多个剂量。因此,单剂量组合物可以含有构成日剂量的此类量或其约数。然而,
应当理解,本公开的化合物和组合物的每日总用量将由主治医师在合理的医学判断范围内决定。
[0174]
同样,本文所述的每种状态、疾病、障碍和病症以及其他状态、疾病、障碍和病症均可以受益于本文所述的组合物和方法。一般而言,治疗状态、疾病、障碍或病症包括预防、逆转或延迟可能患有或倾向于该状态、疾病、障碍或病症但尚未经历或显示其临床或亚临床症状的哺乳动物中的临床症状的出现。治疗还可以包括抑制状态、疾病、障碍或病症,例如,阻止或减少疾病或其至少一种临床或亚临床症状的发展。此外,治疗可以包括缓解疾病,例如,引起状态、疾病、障碍或病症或其临床或亚临床症状中的至少一种的消退。对待治疗的受试者的益处可以是统计学上显著的,或者至少是受试者或医师可感知的。
[0175]
alp功能增强剂的施用可以作为单一事件发生,或者可以在治疗的时间进程内发生。例如,alp功能增强剂可以每日、每周、每两周或每月施用。对于基因疗法,治疗的时间进程通常将为至少一天至几天。某些治疗(例如ert)可能会将治疗从几天延长至几周。例如,治疗可以延长超过一周、两周或三周。对于更多的慢性病症和长期治疗方法,治疗可能从数周延长至数月,或者甚至一年或更长时间。
[0176]
根据本文所述方法的治疗可以在与溶酶体基因中功能丧失变体相关的神经系统疾病、障碍或病症的常规治疗形式之前、同时或之后进行。
[0177]
施用
[0178]
本文所述的药剂和组合物可以根据本文所述的方法以本领域已知的多种方式施用。该药剂和组合物可以在治疗上用作外源性材料或内源性材料。外源性药剂是在体外产生或制造并施用于身体的药剂。内源性药剂是那些在体内通过某种类型的装置(生物的或其他的)产生或制造的用于在体内或向体内的其他器官递送的药剂。
[0179]
如上所讨论,施用可以是颅内、鞘内、胃肠外、肺、口服、局部、皮内、肌内、腹膜内、静脉内、脑室内、皮下、鼻内、硬膜外、眼、颊或直肠施用。
[0180]
本文所述的药剂和组合物可以以本领域熟知的多种方法施用。施用可包括,例如,涉及口服摄入、直接注射(例如,全身或立体定向)、植入经工程改造以分泌目的因子的细胞、药物释放生物材料、聚合物基质、凝胶、可渗透膜、渗透系统、多层包衣、微粒、可植入基质装置、微型渗透泵、可植入泵、可注射凝胶和水凝胶、脂质体、胶束(例如,高达30μm)、纳米球(例如,小于1μm)、微球(例如,1
‑
100μm)、储库装置、上述任何的组合的方法,或其他合适的递送载体,以提供不同比例的所需释放曲线。药剂或组合物的控释递送的其他方法将是技术人员已知的,并且在本公开的范围内。
[0181]
递送系统可以包括例如输注泵,其可以用于以与用于将胰岛素或化疗递送至特定器官或肿瘤的方式类似的方式施用药剂或组合物。典型地,使用此类系统、药剂或组合物可以与生物可降解的、生物相容的聚合物植入物组合施用,该植入物在选定的部位在受控的时间段内释放药剂。聚合物材料的示例包括聚酐、聚原酸酯、聚乙醇酸、聚乳酸、聚乙烯乙酸乙烯酯以及它们的共聚物和组合。此外,控释系统可以放置在治疗靶附近,因此只需要系统剂量的一部分。
[0182]
药剂可以封装在各种载体递送系统中并施用。载体递送系统的示例包括微球、水凝胶、聚合物植入物、智能聚合物载体和脂质体(一般参见,uchegbu和schatzlein编辑(2006)《药物递送中的聚合物(polymers in drug delivery)》,crc,isbn
‑
10:
0849325331)。用于分子或生物分子药剂递送的基于载体的系统可以:提供细胞内递送;定制生物分子/药物释放速率;增加到达其作用位点的生物分子的比例;改善药物向其作用位点的转运;允许与其他药剂或赋形剂的共定位沉积;提高药物在体内的稳定性;通过降低清除率延长药剂在其作用位点的停留时间;减少药剂向非靶组织的非特异性递送;减少由药剂引起的刺激;降低由于药剂的高初始剂量的毒性;改变药剂的免疫原性;减少给药频率,提高产品的口感;或提高产品的保质期。
[0183]
本文所述的利用分子生物学方案的组合物和方法可以根据本领域已知的多种标准技术(参见,例如sambrook和russel(2006),《来自分子克隆的浓缩方案:实验室手册》,冷泉港实验室出版社,isbn
‑
10:0879697717;ausubel等,(2002)《分子生物学中的短协议》,第5版,《当前协议》,isbn
‑
10:0471250929;sambrook和russel(2001),《分子克隆:实验室手册》,第3版,冷泉港实验室出版社,isbn
‑
10:0879695773;elhai,j.和wolk,c.p.1988.《酶学方法》167,747
‑
754;studier(2005)《蛋白质的表达和纯化》41(1),207
–
234;gellissen编辑(2005)《重组蛋白的生产:新型微生物和真核表达系统》,wiley
‑
vch,isbn
‑
10:3527310363;baneyx(2004)《蛋白质表达技术》,taylor&francis,isbn
‑
10:0954523253)。
[0184]
提供本文所述的定义和方法以更好地定义本公开并指导本领域普通技术人员实践本公开。除非另有说明,否则术语应由相关领域的普通技术人员根据常规用法来理解。
[0185]
在一些实施例中,用于描述和要求保护本公开的某些实施例的表达成分的量、性质诸如分子量、反应条件等的数字应被理解为在一些情况下被术语“约”修饰。在一些实施例中,术语“约”用于指示值包括用于确定该值的装置或方法的平均值的标准偏差。在一些实施例中,在书面描述和所附权利要求中阐述的数值参数是近似值,其可以根据由特定实施例寻求获得的期望性质而变化。在一些实施例中,应该根据所报告的有效数字的数量并通过应用普通的舍入技术来解释数字参数。尽管阐述本公开的一些实施例的宽范围的数值范围和参数是近似值,但是在具体实例中阐述的数值被尽可能精确地报告。在本公开的一些实施例中呈现的数值可能包含一定的误差,该误差必然由在它们相应的测试测量值中发现的标准偏差引起。本文中数值范围的叙述仅旨在用作单独提及落入该范围内的每个单独数值的简写方法。除非本文中另有说明,否则每一个单独的值都被并入说明书中,如同其在本文中被单独叙述一样。
[0186]
在一些实施例中,在描述特定实施例的上下文中(特别是在某些所附权利要求的上下文中)使用的术语“一(a和an)”和“该”以及类似的参考可以被解释为覆盖单数和复数两者,除非另外特别指出。在一些实施例中,如本文(包括权利要求)中所用的术语“或”用于意指“和/或”,除非明确指出仅指替代物,或者替代物是互斥的。
[0187]
术语“包含”、“具有”和“包括”是开放式连接动词。这些动词中的一个或多个的任何形式或时态,诸如“包含(comprises、comprising)”、“具有(has、having)”、“包括(includes和including)”,也是开放式的。例如,“包含”、“具有”或“包括”一个或多个步骤的任何方法不限于仅具有那些一个或多个步骤,并且还可以覆盖其他未列出的步骤。类似地,“包含”、“具有”或“包括”一个或多个特征的任何组合物或装置不限于仅具有那些一个或多个特征,并且可以覆盖其他未列出的特征。
[0188]
本文描述的所有方法都可以以任何合适的顺序执行,除非本文另有说明或者上下文以其他方式明显矛盾。关于本文的某些实施例提供的任何和所有实例或示例性语言(例
如,“诸如”)的使用仅旨在更好地说明本公开,而不是对本公开另外要求保护的范围提出限制。说明书中的任何语言都不应当被解释为指示对本公开的实践来说必不可少的任何未要求保护的要素。
[0189]
本文公开的本公开的替代要素或实施例的分组不被解释为限制。每个组成员可以单独提及并要求保护,或者可以与该组的其他成员或本文中存在的其他要素组合提及并要求保护。出于方便或可专利性的原因,组中的一个或多个成员可以被包括在组中或从该组中删除。当发生任何此类包含或删除时,本说明书在本文被认为含有如所修改的组,从而满足所附权利要求中使用的所有马库什组的书面描述。
[0190]
在本技术中引用的所有出版物、专利、专利申请和其他参考文献通过引用以其整体并入本文用于所有目的,其程度如同每个单独的出版物、专利、专利申请或其他参考文献被具体地和单独地指出通过引用以其整体并入本文用于所有目的。本文对参考文献的引用不应被解释为承认这是本公开的现有技术。
[0191]
已经详细地描述了本公开,将明显的是,在不脱离所附权利要求中限定的本公开的范围的情况下,修改、变化和等效实施例是可能的。此外,应当理解,本公开中的所有实例都作为非限制性实例提供。
[0192]
实例
[0193]
提供以下非限制性实例以进一步说明本公开。本领域技术人员应当理解,以下实例中公开的技术代表了发明人已经发现在本公开的实践中很好地发挥作用的方法,并且因此可以被认为构成了其实践的模式的实例。然而,根据本公开,本领域技术人员应当理解,在不脱离本公开的精神和范围的情况下,可以对所公开的特定实施例进行许多改变,并且仍然获得相似或类似的结果。
[0194]
实例1:研究naglu变体对阿尔茨海默病(ad)和帕金森病(pd)病理学的作用
[0195]
本实例描述了体外和体内验证在硫酸乙酰肝素的溶酶体降解中涉及的基因的遗传变异在阿尔茨海默病(ad)和帕金森病(pd)发病机制中的作用。
[0196]
存在令人信服的遗传和生化证据表明,溶酶体功能障碍是诸如阿尔茨海默病(ad)、帕金森病(pd)和额颞叶痴呆(ftd)之类的几种成人发作的神经退行性疾病的常见致病机制。然而,溶酶体功能的年龄依赖性及ad和pd相关联下降背后的遗传变异尚不十分了解。此外,尚未完成每个溶酶体基因的一般群体中的遗传变异对发展ad或pd风险的贡献及其在疾病发病机制中的作用的系统和全面的评估。为了解决目前知识中的这一空白,在ad和pd的病例对照的组群中对45个溶酶体基因进行了单变体和基于基因的分析。如本文所述,发现了与ad和pd两者相关联的几种溶酶体酶基因的变体。这些数据证实了gba与pd的相关性。特别值得关注的是,ad和pd患者中负责硫酸乙酰肝素(hs)代谢的基因(gns、naglu、sgsh和hgsnat)中的罕见功能变体的富集。此外,在来自pd患者的黑质的多巴胺能神经元中存在降低的naglu的转录水平。
[0197]
有趣的是,与年龄匹配的对照相比,naglu转录水平在ad病例中也显著更高,并且在ad的小鼠模型中,随着病理学的发展,naglu转录水平表现出成比例的年龄依赖性增加。由共价连接到特定蛋白核心的hs链组成的硫酸乙酰肝素蛋白聚糖(hspg)是大量的细胞表面和细胞外分子,其与一系列配体相互作用。hspg调控多种致病蛋白(包括淀粉样蛋白(aβ)、载脂蛋白e(apoe)、tau和α
‑
突触核蛋白(α
‑
syn))的寡聚化、清除、内吞和转运。对致病蛋
白的hspg结合的药理学抑制和对hspg合成的遗传性减少促进了致病蛋白的清除并减少了它们的聚集。到目前为止,尚不清楚naglu活性和所产生的hspg积累的降低是否会影响app代谢、aβ斑块负荷或α
‑
syn聚集和扩散。
[0198]
如本文所述,生物化学和基于细胞的测定可以用于全面地表征所选的遗传变体对naglu活性的功能作用。研究了突变型naglu对全长app水平、app转运、神经元中的aβ生成和通过胶质细胞的aβ降解的影响。可以确定naglu的单倍体不足是否会加速在明确表征的ad小鼠模型中存在的ad病理。可以确定在稳定地表达所选变体的naglu缺陷和半合子小鼠的原代神经元中α
‑
syn pff的结合、内化和聚集是否受到影响。最后,在半合子或敲除(ko)naglu小鼠中进行α
‑
syn pff的纹状体内接种,并对psyn聚集体的形成、连通性依赖性扩散及其对疾病进展和寿命的影响进行量化。
[0199]
项目叙述
[0200]
本研究的目的是在体外和体内验证在硫酸乙酰肝素的溶酶体降解中涉及的基因的遗传变异在阿尔茨海默病(ad)和帕金森病(pd)发病机制中的作用。本文所述的研究并入了创新的综合框架,该框架将计算方法和实验数据相结合,以验证所选的naglu变体在体外和体内的功能效应。这里概述的实验可以揭示与ad和pd相关联的新型溶酶体基因,并对ad和pd发病机制中溶酶体功能障碍的机制提供更深入的了解。
[0201]
存在令人信服的遗传学和生物化学证据表明,由溶酶体基因中的纯合突变引起的严重溶酶体功能障碍是几种成人发作的神经退行性疾病(诸如阿尔茨海默病(ad)、帕金森病(pd)和额颞叶痴呆(ftd))的常见致病机制。在ad(5712例/5011名对照)和pd(821例/750名对照)的病例对照的组群中,对45个溶酶体基因进行了单变体和基于基因的分析。在许多与ad和pd两者相关联的溶酶体酶基因中鉴定了变体。重要的是,该数据证实了gba与pd的相关性。特别值得关注的是,ad和pd患者中负责硫酸乙酰肝素(hs)代谢的基因(sgsh、naglu、hgsnat和gns)中罕见的杂合功能变体的富集。在来自pd患者的黑质(sn)的多巴胺能神经元中存在降低的n
‑
乙酰基
‑
α
‑
氨基葡萄糖苷酶(naglu)的转录水平。在ad患者中发现了naglu和sgsh中的预测的罕见杂合功能变体的富集。
[0202]
有趣的是,与年龄匹配的对照相比,naglu转录水平在ad病例中也显著更高,并且在ad的小鼠模型中,随着病理的同时发展,naglu转录水平表现出成比例的年龄依赖性增加。尽管分析确定许多溶酶体酶基因中的单倍体不足是ad和pd的风险因素,但研究了改变的溶酶体hs代谢的影响,并在明确表征的纯合naglu缺陷小鼠中进行了原理论证实验。因此,到目前为止,还没有直接的生物学证据表明naglu的单倍体不足与任何神经系统疾病相关联。由共价连接到特定蛋白核心的hs链组成的硫酸乙酰肝素蛋白聚糖(hspg)是大量的细胞表面和细胞外分子,其与一系列配体相互作用。hspg调控多种致病蛋白(包括淀粉样蛋白(aβ)、载脂蛋白e(apoe)、tau和α
‑
突触核蛋白(α
‑
syn))的寡聚化、清除、内吞和转运。
[0203]
对致病蛋白的hspg结合的药理学抑制和hspg合成的遗传性减少促进了致病蛋白的清除并减少了它们的聚集。大多数hspg和结合的配体被溶酶体蛋白酶、外糖苷酶和硫酸酯酶降解。sgsh、naglu、hgsnat和gns酶参与溶酶体中hs的逐步分解。这些基因中的功能丧失(lof)突变导致溶酶体内部分降解的hs的积累,并且导致粘多糖贮积症(mps)iii型a、b、c和d。尽管hspg的改变的溶酶体降解在多种成人发作的神经退行性疾病的发病机制中的作用尚不明确,但纯合的naglu缺陷小鼠在内侧内嗅皮层(medial entorhinal cortex)中表
现出高磷酸化的tau、aβ和hspg的细胞内积累。此外,mps iiib患者表现出严重的sn神经元丢失,以及颞叶皮层、海马和sn中的神经元中磷酸化的α
‑
syn(psyn)的积累。这些和其他数据强烈地表明,严重的溶酶体功能障碍是ad与pd之间的常见致病机制。然而,如上所述,没有直接的生物学数据表明溶酶体蛋白在成人发作的神经系统疾病中的单倍体不足性(葡糖脑苷脂酶(gba)和帕金森病的一个显著例外)。
[0204]
(i)确定naglu基因中罕见变体的功能作用
[0205]
为了验证其功能作用,用携带3种预测为最有害的变体的慢病毒载体转导来自naglu缺陷小鼠的细胞。测量它们对酶活性和hspg水平的影响。
[0206]
(ii)体外确定naglu基因变体对app代谢、aβ生成和aβ降解的影响,并且体内确定naglu单倍体不足对ad病理的影响
[0207]
测试了来自稳定地表达经验证的变体(如第(i)节所述)的naglu缺陷和半合子小鼠的原代神经元对app转运、app半衰期、app加工机制和aβ生成的影响。测试了来自稳定地表达选定变体的naglu缺陷或半合子小鼠的胶质细胞的aβ摄取和降解。
[0208]
可以确定naglu单倍体不足是否影响5xfad小鼠在早期(4个月)和晚期(8个月)的aβ生成、aβ清除、斑块沉积、突触丢失和神经炎症。使用aav2/9
‑
php.b假型载体在新生半合子小鼠的脑中表达最有害的变体(来自(i)节),并且在不存在fad突变的24月龄小鼠中对naglu单倍体不足性对ad相关表型的影响进行了定量病理学调查。
[0209]
(iii)确定naglu对体外α
‑
syn聚集和体内α
‑
syn扩散的影响
[0210]
假设naglu基因中与pd相关联的功能变体影响α
‑
syn聚集和细胞间传递。可以确定在来自稳定地表达所选变体的naglu缺陷和半合子小鼠的原代神经元中α
‑
syn pff的结合、内化和聚集是否受到影响。最后,可以确定重组酶替代或基因疗法是否能缓解对α
‑
syn pff的内化和聚集的影响。
[0211]
在野生型和表达突变型a53t人α
‑
syn的转基因小鼠中,α
‑
syn pff的纹状体内注射再现了细胞内路易体(lb)病理的积累、sn神经元的选择性丧失和受损的运动协调。α
‑
syn pff的纹状体内接种在半合子或naglu缺陷小鼠中进行,该小鼠在出生时注射了表达最有害的naglu变体的aav2/9
‑
php.b假型载体。对psyn聚集体的形成、连通性依赖性扩散及其对疾病进展和寿命的影响进行了量化。
[0212]
意义
[0213]
ad中的alp功能障碍
[0214]
虽然ad的家族形式在病因上是由增加的淀粉样蛋白β(aβ)生成以及随后aβ在细胞外间隙(isf,间质液)中聚集成可溶性低聚物或不溶性aβ斑块驱动的,但最近对晚发型散发性ad患者的研究表明aβ1的受损的清除。因此,产生与清除之间的平衡决定了aβ水平和发展aβ斑块的倾向。自噬
‑
溶酶体途径(alp)是细胞内细胞器和易聚集蛋白的降解的主要途径。自噬(“自我吞噬”)是一种细胞内降解途径,其负责经由溶酶体消化和回收营养物质。在人脑中的正常衰老期间,alp“核心”基因被转录下调。相反,在ad患者的脑内存在alp的转录上调。在散发性ad脑中,存在beclin 1(一种alp中自噬体形成必需的多功能蛋白)水平的降低、rab5和rab7(调节囊泡沿内体
‑
溶酶体途径转运的小ras相关gtpase(rab)蛋白)水平的升高、大自噬(高lc3
‑
ii水平)和mtor信号传导(磷酸化的p70 s6激酶)的异常激活,以及营养不良神经突中自噬液泡(av)和溶酶体致密体的大量神经元积累。
[0215]
神经病理学研究还已经发现,ad脑中的自噬
‑
溶酶体病理有助于ad的发病机制,然而潜在机制尚不十分清楚。在多种转基因小鼠ad模型中也发现了alp的变化。在两个ad小鼠模型中,beclin 1的单倍体不足性产生对它们的溶酶体的进一步破坏,促进了细胞内和细胞外aβ的积累,并且加剧了神经变性。溶酶体神经氨酸酶1(neu1)的纯合子丢失加剧了ad模型中的aβ病理。相比之下,neu1的过表达降低了ad的病理。这些结果一起表明,alp“核心”基因或溶酶体蛋白的变化加速了ad的病理。细胞研究表明,内体
‑
溶酶体系统是aβ产生的主要位点。然而,关于aβ确切的产生位置,尚无共识。aβ是在体外和体内诱导大自噬后产生的。aβ的积累增加了mtor信号传导,而减少mtor信号传导降低了aβ水平,这表明alp活化与aβ水平之间的负反馈环。在自噬激活下,自噬液泡成为具有最高的γ分泌酶活性的细胞部位。早老素2(psen2)和呆蛋白(nicastrin)(催化必需的γ分泌酶组分)位于溶酶体中。事实上,psen1调节溶酶体ph。
[0216]
体外溶酶体功能的药理学损伤导致aβ产生的变化。溶酶体ph的变化会减少aβ的分泌。溶酶体蛋白酶抑制剂减少淀粉样变性app片段的产生。所有这些研究表明,整体溶酶体功能在正常和异常的aβ前体蛋白(app)加工和随后的淀粉样变性中起重要作用。来自人类病理学、小鼠和细胞模型的所有证据都强烈地表明,自噬诱导中的缺陷发生在疾病早期,但溶酶体清除缺陷发生在疾病的更晚期。
[0217]
pd中的溶酶体功能障碍
[0218]
在人类尸检研究和模型系统中已经很好地确定,内吞转运、溶酶体完整性和溶酶体水解酶活性方面的严重遗传缺陷是突触核蛋白病的风险因素。作为帕金森病(pd)中的致病机制的溶酶体功能障碍由家族性pd中atp13a2(一种溶酶体atp酶)和vps35基因(溶酶体内转运)的突变支持。此外,gba基因(溶酶体水解酶葡糖脑苷脂酶)和smpd1基因(溶酶体酸鞘磷脂酶)的低频变体会增加散发性pd的风险。最近的元分析发现,scarb2(2型溶酶体整合膜蛋白)、tmem175(跨膜蛋白175)、ctsb(溶酶体半胱氨酸蛋白酶组织蛋白酶b)、atp6v0a1(atp酶h+运输v0亚单位a1)和galc(溶酶体半乳糖神经酰胺酶)基因中的常见变体也与pd风险相关联。溶酶体标记物(lamp
‑
1、lamp
‑
2a、组织蛋白酶
‑
d、gba和atp13a2)已经被鉴定为患有散发性pd的患者中lb的组成部分。因此,已经表明,随着疾病的进展,lb和路易神经突(ln)可能在受损的溶酶体周围播散,并且通过溶酶体来源的未降解材料的持续沉积而增大尺寸。
[0219]
尽管机制问题仍然存在,但最近多种基于细胞的模型已经集中在蛋白病变种子的细胞间转移在突触核蛋白病的进展中的可能中心地位上。尚不清楚特异性α
‑
syn菌株是否经由不同的受体或内吞机制被内化。永生化细胞和原代神经元对α
‑
syn的大胞饮摄取似乎是由hspg介导的。然而,尚未评估hs在体内α
‑
syn扩散中的作用。溶酶体加工是原代神经元中的内化的α
‑
syn原纤维的主要命运。简而言之,溶酶体功能的严重药理学扰动导致α
‑
syn原纤维的细胞内加工的畸变,伴随着经由募集内源性α
‑
syn的包涵体形成的增加的速率。控制这种募集的过程仍知之甚少,并且这表明致病物种必须逃脱溶酶体内转运。因此,已经报道,外源性α
‑
syn物种会导致内吞囊泡和溶酶体膜破裂,从而逃脱内吞转运和溶酶体降解。一旦进入胞质溶胶,这些α
‑
syn原纤维或寡聚体可以与可溶性物质相互作用,并启动内源性α
‑
syn的募集。这些结果进一步支持了溶酶体活性和完整性的缺陷可能加速病理性α
‑
syn聚集和传播的观点。
[0220]
溶酶体缺陷被认为有助于α
‑
syn的从头聚集和成熟胞质聚集体的受损的自噬降解。有趣的是,已经报道了使用哺乳动物雷帕霉素(mtor)依赖性或mtor非依赖性自噬增强子靶在几个体外和体内α
‑
syn过表达模型中的神经保护作用。类似地,病毒载体介导的beclin
‑
1表达会减少α
‑
syn转基因小鼠中的α
‑
syn聚集和突触病理。转录因子eb(tfeb)(一种alp的主要激活因子)的过表达也防止α
‑
syn聚集。总体而言,这些研究表明,目的在于恢复pd中的溶酶体功能的新疗法可能提供一种急需的疾病缓解治疗策略。
[0221]
在ad和pd中的硫酸乙酰肝素
[0222]
由共价连接到特定蛋白核心的hs链组成的硫酸乙酰肝素蛋白聚糖(hspg)是大量的细胞表面和细胞外分子,其与一系列配体相互作用。膜hspg充当内吞受体,并且经历组成型和配体诱导的内吞作用。大多数hspg和结合的配体被溶酶体蛋白酶、外糖苷酶和硫酸酯酶降解。hspg调控多种致病蛋白(包括aβ、apoe、tau和α
‑
syn)的寡聚化、清除、内吞和转运。hspg存在于aβ斑块以及lb和ln中。显示,hspg与aβ结合并且加速其寡聚化和聚集。在体外,hs显著刺激α
‑
syn原纤维的形成。hs还介导细胞aβ的摄取。hspg介导α
‑
syn的大胞饮摄取。致病蛋白的hspg结合的药理学抑制和hspg合成的遗传性减少促进了致病蛋白的清除并减少了它们的聚集。这些发现表明,hs和hspg在aβ和α
‑
syn代谢以及ad和pd的发病机制中发挥重要作用。naglu编码n
‑
乙酰
‑
α
‑
氨基葡萄糖苷酶,其参与硫酸乙酰肝素(hs)的溶酶体降解。naglu中的lof突变会导致iiib型粘多糖病(mps
‑
iiib),其也被称为sanfilippo综合征b。在不存在aβ斑块的naglu缺陷和人类mps
‑
iiib患者的脑中,均报告了细胞内全长
‑
app的定性水平升高。与正常对照脑相比,mps
‑
iiib患者呈现出可溶性aβ40水平的显著三倍增加。mps iiib患者呈现出严重的sn神经元丢失,以及磷酸化的α
‑
syn在颞叶皮层、海马和sn中的神经元中的积累。到目前为止,尚不清楚naglu活性和所产生的hspg积累的降低是否会影响app代谢、aβ的产生或清除或α
‑
syn的聚集和扩散。
[0223]
创新
[0224]
本文所述研究的目的是验证遗传学发现,并为ad和pd中的溶酶体功能障碍提供更多见解。此外,本研究可以有助于鉴定患有遗传上确定的溶酶体功能障碍的pd和ad患者,其中此类功能障碍的恢复可以提供有效的疗法。本文概述的研究在系统和全面评估naglu基因中与ad和pd相关联的遗传变异的功能后果方面(第(i)节)是概念性创新的。这些研究将提供对衰老和naglu单倍体不足性对aβ产生和清除(第(ii)节)以及体外和体内α
‑
syn聚集和扩散(第(iii)节)的影响的更好的理解。此外,这些研究仔细检查了神经元病理学,以确定遗传改变naglu的后果及其在临床相关终点对aβ和α
‑
syn病理学的影响。本文所述的研究并入了创新的综合框架,其将计算方法和实验数据相结合,以验证naglu基因在体外和体内的功能效应。基于细胞的测定由生化数据、来自小鼠脑中特定细胞类型的rnaseq数据、人类ad和pd病例及对照中的全基因组基因表达数据以及来自与aβ斑块相关的ad小鼠模型的全基因组基因表达数据补充。第(ii)和(iii)节中所述的研究解决了以下问题:年龄和naglu在易损脑区的单倍体不足性是否影响app加工和转运、aβ斑块负荷和aβ40/42水平以及体外α
‑
syn聚集和体内α
‑
syn扩散。这些研究通过合作创新成为可能,该合作创新涉及具有跨越神经遗传学、溶酶体生物学、lsd动物模型、细胞和小鼠模型中ad和pd病理生理学的专业知识的研究人员。
[0225]
方法
[0226]
这里,最先进的基因组工具可以用于评估与ad和pd风险相关联的naglu遗传变异的体外和体内功能后果。
[0227]
数据和结果
[0228]
溶酶体hs降解基因中的杂合变体影响发展ad的风险
[0229]
在两个病例对照的ad组群中对45个溶酶体基因进行了单变体和基于基因的分析。发现样品由来自667例不相关ad病例和511名对照的全外显子组测序(wes)数据组成。如所预期的,来自exac数据集(欧洲人,非芬兰裔)的基因特异性积累等位基因频率(cmaf)与来自内部ad数据库的cmaf高度一致(r2=0.96)。将罕见蛋白改变变体(cmaf)的负荷与在对照和exac中观察到的负荷进行比较。对于大多数基因,与对照相比,在病例中存在过多的变异,但仅发现与sgsh基因的名义关联(p=4.2
×
10
‑3;优势比(odds ratio)(or)=3.7,95%置信区间(ci)1.4
‑
9.6)。当与exac样品的cmaf相比时,sgsh基因(p=7.9
×
10
‑5;or=3.0,95%ci 1.8
‑
4.9)和naglu基因(p=4.8
×
10
‑4;or=3.7,95%ci 1.4
‑
9.6)通过多项测试校正阈值p<1.0
×
10
‑3(0.05/50)。接下来,使用阿尔茨海默病测序项目(adsp)组群(5045例ad病例和4500名对照)重复这些发现。在该独立样品中复制了naglu(p=3
×
10
‑3;or=2.3,95%ci 1.2
‑
5.2)。值得注意的是,在复制样品中发现的关联是在相同的方向上,并且具有相似的效果大小。
[0230]
随年龄、ad状态和ad小鼠模型中的naglu转录水平
[0231]
来自小鼠的脑细胞类型的rnaseq数据显示,与在神经元中相比,naglu转录物在小胶质细胞中表现出较高的表达水平(约20倍)。在神经病理学正常的人脑样品中,存在naglu转录水平随着年龄的显著增加(p=0.02)(参见,例如图1a)。与年龄匹配的对照相比,在ad病例中naglu转录水平显著更高(p=0.007)(参见,例如图1b)。与野生型小鼠中的水平(参见,例如图1c中的黑线)相比,在ad小鼠模型的皮质中(app,p.k670n/p.m671l/psen1,p.m146v;半合子的[het]或纯合子的[ho];参见,例如图1c),随着ad病理的发展,naglu转录水平也表现出成比例的年龄依赖性升高(参见,例如图1c,右板图)。
[0232]
溶酶体hs降解基因中的杂合变体影响发展pd的风险
[0233]
发现样品由来自ppmi组群的331例不相关pd病例的wes数据组成。来自nfe exac数据集的基因特异性cmaf与来自内部pd数据库的cmaf高度一致(ppmi r2=0.92;内部r2=0.96)。将罕见蛋白改变变体(cmaf)的负荷与在对照和exac中观察到的负荷进行比较。当与exac样品的cmaf比较时,七个基因通过多重检验校正阈值p<1.0
×
10
‑3),包括gba(p=6.1x10
‑6;or=2.1;ci=1.3
‑
3.3),gns(p=2.5x10
‑5;or=2.4;ci=1.4
‑
4.6)和naglu(p=1.0x10
‑4;or=4.7;ci=1.5
‑
8.3)。在hgsnat上也存在趋势(p=8.1x10
‑3;or=1.8;ci=1.1
‑
2.8)。接下来,使用另外的pd组群(wustl)重复这些发现,包括490个pd病例,其中使用人类外显子组芯片获得数据。值得注意的是,在复制样品中发现的关联在相同的方向上并且效应大小相似;naglu(p=3.6x10
‑7;or=3.6;ci=2.8
‑
8.3)和hgsnat(p=9.7x10
‑4;or=1.9;ci=1.4
‑
3.2)。
[0234]
表4.用于功能验证的有害变体
[0235][0236]
具有pd状态的naglu转录水平
[0237]
已经报道了在具有naglu基因突变的mps iiib患者中的sn病理和lb积累。此外,发现与对照相比,在来自pd患者的黑质的多巴胺能(da)神经元中存在naglu基因的转录水平降低(参见,例如图2)。神经元培养物中α
‑
syn pff的制备和使用已在之前被优化。在体外第7天(div),将α
‑
syn pff添加到来自野生型小鼠的原代皮质神经元中。治疗后7天;神经元被固定并用psyn特异性抗体染色。pff诱导内源性表达的α
‑
syn募集到异常的、磷酸化的、不溶性聚集体中(参见,例如图3a和图3b)。α
‑
syn聚集体最初在突触前终末和轴突中表现为小的点状内含物(参见,例如图3a的右下板图)。聚集体生长,并且外观变得更细长和蜿蜒,类似于路易神经突(参见,例如图3a的左下板图)。图3b显示,pbs处理的对照神经元显示出略高于15kda的条带,其对应于单体α
‑
syn。在用pff处理的神经元中出现了几条具有较高分子量的条带。那些额外的条带可能对应于α
‑
syn寡聚体。这是研究溶酶体功能障碍对α
‑
syn聚集的影响的易于处理的体外系统。
[0238]
psyn病理在naglu缺陷小鼠中的传播
[0239]
在六只naglu缺陷小鼠和六只野生型同窝小鼠中进行了α
‑
syn pff或pbs(对照)的纹状体内接种。所有小鼠均在注射后存活,并且目前正在衰老。与已发表的数据一致,注射后30天(dpi),pbs治疗的动物未表现出psyn病理(参见,例如图4a)。相反,pff注射的野生型小鼠表现出丰富的同侧psyn病理,以及非常少的对侧psyn病理(参见,例如图4b)。在90dpi时,在pff注射的野生型小鼠中存在psyn病理梯度,其中同侧强度大于对侧强度。这种梯度在运动皮质和sn中非常明显。在杏仁核和体感皮质中存在更对称的病理。令人惊讶的是,用α
‑
syn pff治疗的naglu缺陷小鼠在30dpi时在注射部位的同侧和对侧两者的前额叶和外嗅皮质中均表现出α
‑
syn病理(参见,例如图4c)。naglu缺陷小鼠似乎具有更对称的psyn病理,这可能表明向对侧的扩散比在wt小鼠中观察到的要多。目前正在分析更多的α
‑
syn pff治疗的小鼠,以进一步表征naglu缺陷对psyn病理的区域和时间扩散的影响,并扩展这些表明naglu缺陷小鼠中psyn病理的扩散增加的初步结果。
[0240]
研究设计与方法
[0241]
(i)确定naglu基因中变体的功能效应
[0242]
评估对蛋白产品的影响
[0243]
评估与所鉴定的ad或pd相关联的所有naglu基因的变体超出了本文所述研究的范围。因此,本研究集中于在naglu基因中鉴定的3
‑
5个顶级变体。基于ad/pd患者中的频率、通过sift和polyphen2预测的对蛋白的影响以及gerp保守性评分来定义顶级变体。基于来自发现和复制样品的数据强度来选择这些基因。可以确定naglu基因中所选变体对酶活性、蛋白水平和溶酶体功能的影响,其中表4中所列的3种变体是重点领域。已经使用非常严格的标准来选择naglu中的潜在功能变体(之前被确定为致病性变体的变体)。然而,表征它们对蛋白水平和酶活性的影响非常重要。来自naglu缺陷小鼠的细胞已经被永生化。表4中概述
的变体被转导,并且可以确定其对酶活性和蛋白水平的影响。还可以确定对溶酶体功能和hspg的积累的影响。使用定点诱变对所选的变体进行工程改造,并将所选的变体亚克隆到如先前所述的慢病毒载体中。慢病毒载体按照涉及重组或合成核酸分子的nih研究指南的第iii
‑
e
‑
1节在bsl2设施中生产、处理和处置。为了确定变体对酶活性的影响,如前所述使用荧光法测定naglu活性。如前所述,确定变体以影响溶酶体功能。通过elisa来定量hspg的水平。为了确保严谨性和再现性,在至少3个独立实验中由双盲观察者一式三份进行定量。
[0244]
预期的结果
[0245]
预期naglu变体会导致功能的部分丧失,增加溶酶体中部分降解的hs,并改变alp功能。预期检测到5
‑
20%的残留naglu活性。如果所选的变体与野生型水平相比未能降低活性,则评估其对亚细胞定位和错折叠的影响。可以选择另外的与ad或pd相关联的变体,以测试其对酶活性的影响。
[0246]
(ii)(a)体外确定naglu对app代谢、aβ生成和aβ降解的功能作用
[0247]
评估对app转运、内吞作用和亚细胞定位的影响
[0248]
多项研究已经表明,app的内吞作用对于其在app淀粉样变途径中与内体和多囊体内的β分泌酶和γ分泌酶共同定位是至关重要的。继发于溶酶体功能障碍的内体通量的损伤导致在该细胞器内的增加的转运时间,这增加了β裂解和γ裂解的倾向,并且因此增加了aβ的生成。在没有aβ斑块的naglu缺陷和sanfilippo b两者患者的脑中已经报告了升高的细胞内全长app水平。为了确定naglu基因中的选定变体是否影响app的稳态水平、app内吞作用或app进入溶酶体用于降解的增强的通量,可以使用如先前公布的细胞表面生物素化测定法来确定细胞内app出现的动力学和细胞表面中app的水平。在用蛋白合成抑制剂环己酰亚胺处理0、5、10、30分钟时,通过蛋白免疫印迹来测量对全长app半衰期的影响。共定位技术用于研究对app和sorl1亚细胞定位的影响。分别通过蛋白免疫印迹和rt
‑
qpcr来测量app加工机制的蛋白和转录水平,包括α分泌酶(adam10和adam17)、β分泌酶1(bace1)和γ分泌酶复合物(psen1和呆蛋白)。
[0249]
评估对aβ生成的影响
[0250]
很大一部分app被靶向溶酶体,并且在存在溶酶体酸化抑制剂的情况下,app水平在细胞中迅速升高,这表明溶酶体降解会驱动app蛋白水解,以阻止aβ肽的形成。与正常对照脑相比,sanfilippo b患者呈现出可溶性aβ水平的显著升高(3倍)。据报告,在naglu缺陷小鼠的脑中,aβ寡聚体水平显著升高。这些发现表明,在具有naglu缺陷的小鼠和人类两者中,hs的积累和溶酶体功能障碍导致γ分泌酶依赖性异常app加工。因此,可以评估所选的变体是否影响细胞培养物中aβ的生成。如先前所述进行原代神经元培养。在神经元特异性启动子(突触素)下携带选定变体和野生型的慢病毒载体用于转导来自naglu缺陷和半合子两者小鼠的原代神经元。使用不同体积的浓缩慢病毒来确定神经元中的适当表达水平。通过rt
‑
qpcr和荧光测定法来测量naglu水平。通过如先前公布的夹心elisa来检测细胞裂解物和细胞培养基中的aβ物种。简而言之,用小鼠单克隆包被抗体hj2(抗aβ35
‑
40)和hj7.4(抗aβ37
‑
42)捕获aβx
‑
40肽和aβx
‑
42肽。hj5.1(抗aβ13
‑
28)(一种靶向中心结构域的生物素化抗体)或hj3.5(其靶向n
‑
末端氨基酸)被用作检测抗体,然后是链霉亲和素
‑
聚
‑
hrp
‑
40。通过蛋白免疫印迹来测量app衍生的蛋白水解片段,诸如α
‑
和β
‑
ctf以及sappα和sappβ。如前所述,通过蛋白免疫印迹来监测全长app的水平。
[0251]
评估对aβ降解的影响
[0252]
小胶质细胞在aβ斑块周围增殖,并且吞噬aβ材料,但随后的降解受损,导致ad中进行性aβ积累。小胶质细胞为何可以摄取原纤维aβ但不能降解原纤维aβ尚不清楚。然而,来自ad患者的小胶质细胞显示beclin
‑
1的减少和随后的alp功能障碍。此外,不溶性原纤维aβ影响氯离子通道cic
‑
7向原发性小胶质细胞中的溶酶体的转运,这损害溶酶体降解。然而,恢复溶酶体酸化会增强aβ的降解。总之,该证据表明小胶质细胞中的alp不足性可能有助于ad发病机制。这里介绍的数据挖掘工作揭示了,与在神经元中相比,naglu在小胶质细胞中呈现出较高的表达水平。因此,可以评估来自用选定的变体转导的naglu缺陷和半合子小鼠的原代小胶质细胞是否可以摄取和降解外源性aβ。aβ的摄取和降解评估如先前公布的进行。
[0253]
评估对alp功能的影响
[0254]
在naglu缺陷小鼠的心脏和脑组织中增加的beclin1、p62和lc3
‑
ii水平表明溶酶体自噬系统的异常活性,并伴有自噬体的积累。可以评估半合子naglu小鼠的神经元是否表现出alp功能障碍,以及这些变化是否通过所选的变体而增加。lc3和p62的蛋白免疫印迹可以用作巨噬细胞活化的间接指标。在不存在或存在自噬系统的激活剂(雷帕霉素和torin1)、自噬抑制剂(巴弗洛霉素a1)、溶酶体营养剂(氯喹、氯化铵)和e64/亮抑蛋白酶肽(如前所公布的)的情况下,通过细胞中存在的lc3
‑
ii的量来评估自噬通量。通过使用mcherry
‑
gfp
‑
lc3标记物的活细胞成像对此进行补充。可以通过lc3和lamp1的共定位来进一步评估自噬体
‑
溶酶体融合。溶酶体跟踪器(lysotracker)用于定量每个细胞的酸性隔室数。tfeb的活化通过其核定位来评估。rt
‑
qpcr用于测量tfeb调节的mrna转录物(sqstm1/p62、map1lc3b和lamp2)的转录水平的变化。为确保严谨性和再现性,研究者在定量和分析阶段期间对基因型视而不见。对于每个实验,在所述的每个组中对获得的数据进行平均。实验以一式三份进行,每个基因型至少使用两种独立产生的制剂。使用双向anova和适当的事后检验来检验具有统计学意义的差异,以确定每种标记物或功能分析是否与naglu相对于对照相关。
[0255]
aav2/9
‑
php.b载体的生成
[0256]
naglu野生型和最有害的变体被亚克隆到aav2/9
‑
php.b载体中。aav2/9
‑
php.b以比aav9大至少40倍的效率在整个中枢神经系统(cns)中转移基因,并跨多个cns区域转导大多数星形胶质细胞和神经元。从unc病毒载体核心机构(unc viral vector core facility)获得了高滴度的aav2/9载体储备液。对于本项目中概述的所有实验,在乳酸林格氏溶液中将aav载体储备液稀释至1012vg/ml。
[0257]
预期的结果
[0258]
预期naglu对app转运、app代谢、aβ生成或aβ降解存在基因剂量效应。此外,预期本文概述的实验允许评估naglu基因中选定变体对神经元和小胶质细胞存活的影响。可替代地,可以使用5xfad转基因小鼠的原代神经元或n2a695细胞,并用naglu基因中的选定变体进行转导,并且评估对aβ生成的影响。在体外鉴定出naglu基因中影响ad的风险和发病机制的变体后,下一步是利用直接从人成纤维细胞产生诱导的多能干细胞(ipsc)和基因组编辑方法的进展。来自携带naglu基因中的变体的ad患者的ipsc衍生的神经元或胶质细胞可以用于比较此类变体与等基因crispr校正细胞相比对app代谢的影响。结合来自本实例的结果和naglu活性的荧光测定法的可用性,可以筛选ad病例和对照的脑脊液(csf)、血浆、血清
或脑组织,以检测可以用作ad的生物标记物的特定缺陷。
[0259]
(ii)(b)确定naglu单倍体不足性对老年小鼠中ad病理的发展的功能性影响
[0260]
虽然已经表征了naglu在小鼠和人类中完全功能丧失的神经退行性后果,但对该基因的单个拷贝(单倍体不足性)的长期后果的了解非常少。一直以来,人们都认为半合子小鼠和人类是正常的。然而,最近表明,溶酶体基因中的单倍体不足性会导致在人和小鼠中的显著的代谢异常。这里假设ad病理由较温和形式的遗传性alp功能障碍发展而来,并且其出现可能需要额外的年龄相关alp损伤。主要终点为在4个月大时(在斑块沉积之前)测得的aβ水平和在8个月大时在小鼠中存在引起fad的突变的斑块负荷。对aβ水平的影响是表达与ad相关联的最有害naglu变体的24月龄naglu半合子小鼠中的主要终点。
[0261]
对ad病理的小鼠模型的影响
[0262]
在患有sanfilippo b病的人类患者中,naglu蛋白的完全功能丧失导致可溶性aβ40的水平比正常对照脑显著升高三倍。在ad小鼠模型的皮质中,随着ad病理的发展,naglu转录水平表现出成比例的年龄依赖性增加(参见,例如图1c)。与ad转基因小鼠一样,人类认知能力下降与aβ斑块负荷不成比例,但与可溶性aβ物种相关。考虑到来自人sanfilippo b患者和naglu缺陷小鼠的数据支持这些基因在细胞内aβ生成中的作用,可以确定在良好表征的ad小鼠模型中,轻度溶酶体损伤(naglu中的半合子)是否会加速aβ生成,该ad小鼠模型携带有利于aβ生成的家族性阿尔茨海默病(fad)突变。naglu缺陷小鼠表现出高度硫酸化的hs脑积累、神经炎症、神经元和小胶质细胞中的溶酶体增大以及约4mo的突触蛋白减少,随后是昼夜节律改变、听力和视力缺陷,以及在老年小鼠(>8mo)中,表现出浦肯野细胞的丧失和运动协调受损。naglu缺陷小鼠的中位寿命为约12mo龄。5xfad模型是一种极具侵袭性的aβ沉积模型,其在1.5个月时出现神经内aβ42,在2个月时出现斑块,在4个月时出现突触标记物和记忆缺陷的丢失,并且在9个月龄时出现神经元丢失。斑块的发展伴有反应性胶质增生。为了进一步确定naglu单倍体不足性是否会加剧现有的淀粉样变性过程,将naglu小鼠与5xfad转基因小鼠杂交。naglu和5xfad小鼠在c57bl/6背景下是同源的。如前所述,通过组织学来确定最有害的naglu变体(在第(i)节中描述)和naglu的基因剂量对aβ斑块负荷的影响,并通过夹心elisa来确定第4个月和第8个月的aβ40/aβ42水平。通过经由蛋白免疫印迹测量app
‑
ctf来评估app代谢。在5xfad小鼠中,四个月是aβ斑块沉积的早期时间点,以便检测aβ积累是否开始得更早,而八个月代表当aβ斑块丰富时的晚期。
[0263]
随机化、生物学变量和样品量
[0264]
样品量计算表明,至少需要n=10只小鼠/组来检测斑块负荷的40%增加(sd=20%,α=5%)以及具有80%功效的洗涤剂可溶性和不溶性aβ40和aβ42。在出生时使用aav2/9
‑
php.b向二十(20)只naglu缺陷小鼠和20只与5xfad杂交的naglu半合子小鼠注射最有害的naglu变体。此外,还采集了4个月龄和8个月龄的20只naglu缺陷小鼠、20只与5xfad杂交的naglu半合子小鼠以及20只另外的5xfad小鼠(以对照遗传背景),用于组织学研究和生物化学研究。由于雌性通常比雄性具有更大的aβ积累,因此每组由10只雄性和10只同窝雌性组成(n=20)。一旦为每个实验生成了实验动物,实验室的独立成员就随机为每只动物分配数字。因此,与本研究关系最密切的研究人员不了解基因型和治疗方案,从而确保无偏倚的实验。用于生化分析和组织学分析的样品保留相同的随机分配的编号。通过在一次实验中使用同源动物和同一批试剂,将生物变量保持在最低限度。
[0265]
naglu单倍体不足性对老年小鼠的影响
[0266]
ad病理(例如aβ斑块)通常是年龄依赖性的。然而,已发表的研究尚未解决年龄与alp功能障碍之间的相互作用。大多数在体内评估alp在ad中作用的研究已经使用了药理学方法,或者完全没有alp基因和短期终点。在10个月大的naglu缺陷小鼠的大脑中,已报告了aβ寡聚体的增加。如本文所述,可以采用遗传学方法以用于降低naglu的内源性水平,并且可以对遗传性慢性溶酶体损伤对涉及aβ的ad相关表型的影响进行定量病理学调查。结果显示,在正常人脑样品中,naglu转录水平随着年龄的增加而高度显著增加(参见,例如图1a)。此外,与年龄匹配的对照相比,在ad病例中naglu转录水平显著更高(参见,例如图1b)。这些结果表明,对来自naglu的聚集蛋白的代偿性应答可能是正常衰老过程的一部分。在ad模型中发现的异常升高表明,它们试图控制aβ的异常水平。因此,naglu中的单倍体不足性可能会加剧老年小鼠中的ad相关表型。
[0267]
样品量
[0268]
样品量计算表明,至少需要n=15只小鼠/组来检测具有80%功效的洗涤剂可溶性和不溶性aβ40和aβ42的20%增加。在出生后第1
‑
2天,使用aav2/9
‑
php.b向十五(15)只naglu缺陷小鼠、15只naglu半合子小鼠和15只野生型小鼠注射最有害的naglu变体。允许小鼠恢复并存活至至少12个月大。将垂死的小鼠麻醉并处死,以收集脑生化研究结果,诸如洗涤剂可溶性和不溶性aβ40和aβ42水平。
[0269]
aβ斑块定量和aβ生成
[0270]
固定的冷冻脑切片(50μm)在小鼠的亚组群中用x
‑
34进行染色,并用hj3.4(抗aβ)抗体进行免疫染色,以量化斑块负荷(以%面积表示)。将来自对侧半球的脑组织匀浆中的aβ水平分级为可溶性(pbs)和不溶性(5m胍)级分,并使用elisa进行定量。评估了对app加工机制的影响。
[0271]
突触标记物
[0272]
突触丢失是人类和ad小鼠模型中的常见发现。可以通过蛋白免疫印迹(使用针对突触前标记物:snap
‑
25、囊泡相关膜蛋白2、突触结合蛋白1和突触素的抗体,如前所述)来评估naglu单倍体不足性是否会加速5xfad小鼠中的突触丢失。
[0273]
体内aβ微透析
[0274]
aβ在大脑中具有相对短的半衰期,其中在小鼠间质液(isf)中为约1
‑
2小时并且在人脑脊液(csf)中为约8小时。为了研究naglu对aβ生成和清除的影响,使用体内微透析来动态地评估在3
‑
4个月大时注射avv2/9或pbs的5xfad/naglu(+/
‑
)小鼠、5xfad/naglu(
‑
/
‑
)小鼠和5xfad同窝小鼠的海马中的isf aβ代谢。为了评估清醒、自由活动小鼠的海马中随时间推移的isf aβ水平,如前所述进行了体内微透析。简而言之,在异氟醚麻醉下,立体定向地将引导套管植入到海马上方(前囟后3.1mm,中线旁2.5mm,和硬脑膜下1.2mm,呈12
°
角)。微透析探针通过引导套管被插入到大脑中。人工csf被用作微透析灌注缓冲液。每60
‑
90分钟采集微透析样品,并通过elisa评估aβ40或aβ42。在6小时内的aβ的平均浓度被定义为isf aβ的基础浓度。对于每只动物,将所有aβ浓度归一化为该小鼠的基础aβ浓度。在确定基础浓度后,向小鼠施用血脑通透性γ
‑
分泌酶抑制剂(ly411575,3mg/kg皮下注射),以快速阻断aβ的产生。每60分钟采集微透析样品,持续6小时,并且然后通过elisa测定aβ40。根据aβ的变化%相对于时间的半对数图斜率来计算isf aβ的半衰期。在半衰期分析中仅包括持续降低
的aβ值。基于功效分析,n=10只小鼠/组检测到isf aβ水平和清除率的30%降低。(5组
×
10=50只小鼠;雌性和雄性数目相等)
[0275]
alp功能障碍
[0276]
如上所述,用抗lamp1、lc3和p62抗体对来自与5xfad小鼠杂交的naglu缺陷、naglu半合子和naglu半合子小鼠的脑切片进行免疫染色。
[0277]
对神经营养不良和反应性胶质增生的影响
[0278]
发明人的先前研究表明,naglu缺陷小鼠表现出增加的星形胶质增生。因此,在平行研究中,脑切片用抗cd11b和抗gfap抗体染色,以检查所选基因的一个单个拷贝对反应性胶质增生的影响。使用网状蛋白
‑
3(rtn
‑
3)抗体(rtn
‑
3选择性地积累在营养不良的神经突中)对固定的冷冻脑切片进行免疫染色,以定量营养不良的神经突,如前所做。
[0279]
预期的结果
[0280]
预期对于naglu基因半合子的小鼠会加速并加重5xfad小鼠中的aβ斑块负荷。预期naglu单倍体不足性会影响老年小鼠中的app代谢和aβ生成,随后会增加突触丢失和反应性胶质增生,而不会生成aβ斑块。如果在半合子小鼠的大脑中未发现app代谢和aβ生成的变化,则可以通过注射携带针对它们的经验证的shrna/rnai的aav2/9载体,在新生的5xfad转基因小鼠中敲除naglu的转录物。crispr技术可以用于在选定的基因中产生对体外测定影响最大的变体的敲入小鼠。为了扩展在naglu小鼠中的发现,可以将相同的方法应用于有小鼠模型可用的降解hs的其他溶酶体酶(例如,n
‑
磺基葡糖胺磺基水解酶[sgsh(jax 003780)])。
[0281]
(iii)确定naglu对体外α
‑
syn聚集和体内α
‑
syn扩散的影响
[0282]
在体外确定naglu对α
‑
突触核蛋白的摄取、转运、聚集和清除的功能作用
[0283]
永生化细胞和原代神经元对α
‑
syn的大胞饮摄取似乎是由hspg介导的。此外,hs在体外显著地刺激α
‑
syn原纤维的形成。已经在培养的神经元中建立了良好表征的α
‑
syn聚集模型。在该模型中,将由重组α
‑
syn生成的pff直接添加到原代神经元中,并被神经元内吞。这些pff诱导内源性表达的α
‑
syn募集为异常的、磷酸化的、不溶性和泛素化的聚集体。体外来源于野生型的非转基因小鼠的原代神经元中由内源性α
‑
syn形成这些聚集体遵循2
‑
3天的滞后期,随后在轴突中在第4
‑
7天形成,在第7
‑
10天扩散至体细胞树突状隔室中,并在加入pff后约14天神经元死亡。使用这一良好表征的模型,可以确定naglu对α
‑
syn原纤维的摄取、转运、聚集和清除的影响。还可以确定在来自用最有害的naglu变体转导的naglu缺陷和半合子小鼠的原代海马神经元中内源性α
‑
syn的清除率是否存在任何变化。可以确定在用α
‑
syn pff治疗后(参见,例如图2)在来自naglu缺陷小鼠的原代神经元中α
‑
syn聚集体(内含物)的速率和水平。最后,可以确定重组酶替代是否可以挽救来自naglu缺陷小鼠的神经元中的α
‑
syn pff。
[0284]
α
‑
syn pff的产生
[0285]
从细菌中制备重组单体α
‑
syn,并根据先前建立的方案通过尺寸排阻和离子交换色谱法依次纯化。通过在37℃下搅动重组单体持续约72
‑
120小时,然后使用具有指定分子量截止参数的离心过滤装置进行尺寸选择,来制备原纤维形式的α
‑
syn。用于生成pff的条件已经被优化。pff在tris缓冲的nacl中稀释,并且在5
‑
10div后加入到培养的原代神经元中。暴露4
‑
7天后,通过免疫荧光或顺序提取和免疫印迹来确认pff转导和接种。经由用抗
psyn(ser129)抗体、克隆81a(biolegend,mms
‑
5091)免疫荧光并且通过蛋白免疫印迹来检测来自内源性α
‑
syn的异常α
‑
syn聚集体。α
‑
syn pff在bsl2设施中生产、处理和处置。
[0286]
α
‑
syn pff转运、内吞作用和亚细胞定位
[0287]
溶酶体加工是原代神经元中内吞的α
‑
syn原纤维的主要命运。用突触前(cspα)、内吞(eea1)、自噬体(rab7)和溶酶体(lamp1)标记物对pff处理的神经元进行共染色,以确定在内溶酶体途径中α
‑
syn聚集体的转运是否存在变化。
[0288]
评估对alp功能的影响
[0289]
α
‑
syn聚集体通过降低自噬体清除率而损害整体巨噬细胞。因此,可以确定对自噬途径的药理学调节是否能改善α
‑
syn清除率。
[0290]
评估对伴侣介导的自噬(cma)的影响
[0291]
α
‑
syn被伴侣介导的自噬(cma)降解,并且易于聚集的α
‑
syn突变体阻断cma。因此,检查lamp2a和hsp70蛋白水平,以了解它们在pff处理的细胞中是否受到影响。用cma激活剂处理pff处理的神经元;在细胞裂解液和条件培养基中确定ar7(视黄酸受体α特异性拮抗剂)和α
‑
syn水平。
[0292]
对内吞和溶酶体膜完整性的影响
[0293]
α
‑
syn pff诱导内吞后囊泡和溶酶体破裂。这些破裂的囊泡对eea1、lc3和半乳糖凝集素
‑
3呈阳性。通过gal
‑
3和lc3染色可以确定α
‑
syn pff是否影响溶酶体膜的完整性。
[0294]
用酶替代的挽救实验
[0295]
在存在或不存在摄取/结合抑制剂(甘露糖
‑6‑
磷酸:m3655,sigma)的情况下,在暴露于α
‑
syn pff之前、期间和之后添加重组naglu,并检测对α
‑
syn pff聚集的影响。已经存在足够的重组naglu用于在体外添加到缺陷神经元中。
[0296]
预期的结果
[0297]
预期naglu在体外对α
‑
syn pff的摄取、转运和聚集的基因剂量效应和重组酶可以挽救那些效应。naglu缺陷细胞在细胞膜和内溶酶体系统中积累hs和hspg。因此,预期α
‑
syn pff的摄取增加,随后内吞囊泡和溶酶体膜破裂,这将增加胞质水平和内源性α
‑
syn的募集。可以生成慢病毒载体,并且易于聚集的α
‑
syn突变体可以在来自naglu缺陷和半合子小鼠的原代神经元中过表达,并且评估了聚集和降解的速率。可替代地,可以使用来自过表达人a53tα
‑
syn转基因小鼠的原代神经元,并且可以用naglu基因中的选定变体对它们进行转导,并且随后测试对α
‑
syn的影响。来自携带naglu基因中的变体的pd患者的ipsc源性神经元也可以用于比较此类变体与等基因crispr校正的细胞相比对α
‑
syn加工的影响。
[0298]
确定naglu对体内α
‑
syn扩散的影响
[0299]
积累的实验数据表明,α
‑
syn的细胞间传递遵循与针对朊病毒蛋白观察到的接种原则类似的接种原则。向过表达人a53tα
‑
syn的年轻小鼠(约3
‑
4月龄)中脑内注射含有聚集的α
‑
syn的脑提取物(来自路易体病的病例的尸检脑提取物)会刺激在宿主中形成psyn损伤,早在注射后30天(dpi)被观察到;到约90dpi,psyn在脑的解剖连接区域中广泛且丰富,这表明与在人类pd病例中观察到的α
‑
syn沉积的明显扩散类似。在100dpi左右,与未注射的小鼠相比,小鼠出现运动功能障碍和过早死亡(约126dpi)。在非转基因(野生型)宿主小鼠中,脑内注射合成(人或小鼠)α
‑
syn pff也诱导了lb样病理和神经元变性(参见,例如图4b)。大约有50%的注射有具有lb脑的痴呆的不溶性psyn的野生型小鼠发展了psyn病理。相
比之下,由人和小鼠α
‑
syn pff诱导psyn病理的效率分别为90%和100%。在30dpi时,psyn阳性的lb样积累仅在注射部位同侧(参见,例如图4b和图4c)。在受影响的同侧区域内以及在对侧新皮质内的lb/ln病理学显示在90和180dpi检查的小鼠中,psyn免疫反应性显著增加。在注射pff后,sn致密部(snpc)α
‑
syn病理逐渐发展,从30dpi时的淡细胞质积累演变为90和180dpi时的致密核周lb样包涵体,尤其是在腹内侧snpc群体中。snpc多巴胺能(da)神经元在90和180dpi时分别伴随减少15%和35%,表明lb/ln在snpc da神经元丢失之前形成。因此,lb/ln的传播是连接性依赖的,并且病理性α
‑
syn的积累似乎是在snpc da神经元丢失的上游并与snpc da神经元丢失直接相关。这种纹状体内注射α
‑
syn pff的体内模型概括了细胞内lb/ln病理的积累、snpc da神经元的选择性丢失和受损的运动协调性。lb和ln都含有hspg。然而,hs在体内α
‑
syn扩散中的作用尚未得到评估。到目前为止,还不清楚naglu活性和所产生的hspg积累的降低是否影响α
‑
syn的聚集和扩散。如本文所述,表达与pd相关联的最有害naglu变体的naglu缺陷和半合子小鼠可以用于测试hs和hspg积累是否影响α
‑
syn聚集、扩散和加速体内疾病。
[0300]
α
‑
syn pff的纹状体内接种
[0301]
如上所述来制备α
‑
syn pff。在出生时使用aav2/9
‑
php.b在注射了最有害的naglu变体的naglu缺陷和半合子小鼠以及naglu缺陷小鼠、半合子小鼠和十二只野生型小鼠中,对α
‑
syn pff进行纹状体内接种。量化了psyn聚集体的扩散,并且评估了α
‑
syn pff是否会影响naglu小鼠的寿命。在单次接种小鼠αsyn pff(或作为对照的pbs或αsyn单体)的情况下,对3
‑
4月龄野生型、naglu缺陷或半合子小鼠在背侧纹状体(0.2mm a/p,相对于前囟2.0mm m/l,颅骨表面下3.2mm)中单侧注射。允许小鼠恢复并在注射后年龄至30、90或180天,此时收获大脑并用抗psyn(ser129)抗体进行免疫组织化学处理。使用抗泛素和hsp90进行psyn共定位染色。根据功效分析,n=12只小鼠/组检测到psyn水平的30%增加。(5组x12=60只小鼠)
[0302]
αsyn和psyn的定量
[0303]
从每只动物的半脑中解剖皮质、海马、纹状体和脑干,并且通过顺序去污剂提取来分离不溶性αsyn,然后使用抗突触核蛋白
‑
1/克隆42(bd biosciences)和抗psyn 81a(biolegend)作为捕获抗体进行elisa和蛋白免疫印迹(如前所公布的)。
[0304]
使用先前公布的针对酪氨酸羟化酶(th)和多巴胺转运体(dat)的抗体,对来自pff治疗的动物和pbs治疗的动物的同侧和对侧纹状体进行免疫印迹分析。评估sn中的脑萎缩和神经元丢失,并如上所述进行损伤和炎症的标记物(例如gfap,iba
‑
1)的免疫组织化学。
[0305]
比较pff治疗的naglu缺陷动物与pbs治疗的naglu缺陷动物之间的寿命。已知在野生型小鼠中,α
‑
syn pff注射6个月后,旋转棒和悬丝测试对于检测运动障碍是最灵敏的。之前已经在小鼠模型中公布了旋转棒和线悬挂行为测试的使用,并且已知适当的实验设计和统计工具(用于多组比较的具有事后分析的anova,以及用于成对比较的学生t检验)。在pff治疗的动物和pbs治疗的动物中进行步态分析。之前的研究已经表明,在lsd的小鼠模型中,步态分析对捕捉“帕金森样”体征非常敏感。
[0306]
关于旋转棒和悬丝测定的性能,对于一次五组之间的比较,当正常动物在60秒下进行并且naglu缺陷动物在0秒(40周)下进行,并且标准偏差为30秒时,效应大小为0.89。在0.89的效应大小,α=.05和功效=.95的情况下,需要6只动物就能够检测到组之间的显著
差异。关于寿命,对于中位寿命为约730天的正常动物和半合子动物与中位寿命为约322天的naglu缺陷动物且标准差为20天的五个组之间的比较,效应大小为5.51。在5.51的效应大小,α=.05和功效=.95的情况下,仅需要2
‑
3只动物就能够检测到组之间的显著差异。由于使用了10
‑
12只小鼠/组,因此行为和寿命研究具有足够的效力。
[0307]
预期的结果
[0308]
预期α
‑
syn pff注射会加速naglu缺陷小鼠的表型,增加胶质增生、神经变性,使运动障碍恶化和使寿命缩短。半合子naglu小鼠使psyn病理的繁殖恶化。可替代地,可以生成携带α
‑
syn突变的aav2/9载体,并立体定向地注射到年轻naglu缺陷小鼠的纹状体中,或者可以将过表达的人a53tα
‑
syn小鼠杂交到naglu缺陷小鼠中,并评估psyn病理、疾病进展和寿命的变化。同样的方法可以应用于sgsh缺陷小鼠。
[0309]
实例2:研究自噬
‑
溶酶体途径(alp)中年龄依赖性和阿尔茨海默病(ad)相关联下降背后的遗传变异
[0310]
本实例描述了阿尔茨海默病(ad)发病机制中涉及的自噬
‑
溶酶体途径(alp)的功能障碍背后的遗传变异的鉴定。
[0311]
尽管多项体外和体内研究表明自噬溶酶体途径(alp)功能障碍有助于ad的发病机制,但尚不完全了解alp功能的年龄依赖性和与ad相关联的下降背后的遗传变异。导致ad的基因和多个ad风险基因中罕见的功能变体会导致alp功能障碍。对分离的群体中单个alp基因的研究支持ad与溶酶体贮积症(lsd)之间的遗传重叠。然而,尚未完成对alp中每个基因的一般群体内的遗传变异对发展ad的风险的贡献及其在ad发病机制中的作用的系统和全面的评估。为了解决当前知识中的这一空白,本文描述的研究可以鉴定和优先考虑在alp的基因中具有较大的效应量的与发展ad风险相关联的罕见功能变异。如本文所述,耦合计算方法和实验数据的整合框架可以用于在体外和体内验证所选alp基因的功能效应。首先,将来自33,350名非芬兰欧洲对照的alp的各基因中罕见功能变体的负荷与2,000例ad病例和3,000名对照进行比较。对额外独立样品(包括2,000例ad病例和2,000名对照)的结果与来自阿尔茨海默病测序项目(adsp)的10,000份公开可获得的样品一起进行了复制。其次,使用基于细胞的测定来检查与ad风险相关联的候选alp基因中所选变体对酶活性、蛋白稳定性和/或mrna水平的影响及其对溶酶体功能的影响。检查了经验证的功能变体对淀粉样变性和aβ降解的影响。最后,在与ad风险相关联的候选alp基因的半合子或敲除模型中,对ad病理的自发发展进行了定量和定性病理学调查。还在良好表征的ad小鼠模型中测量了候选alp基因的基因剂量对aβ斑块负荷的影响。本文概述的研究可以揭示与ad相关联的新alp基因。这些实验可以对ad发病机制中alp功能障碍的机制提供更深入的了解,并为将目前针对溶酶体贮积症的治疗策略重新用于ad的潜在治疗奠定基础。
[0312]
本文所述研究的目的是鉴定阿尔茨海默病(ad)发病机制中涉及的自噬
‑
溶酶体途径(alp)的功能障碍背后的遗传变异。这些研究并入了创新的整合框架,该整合框架将计算方法和实验数据相结合,以在体外和体内验证选定alp基因的功能效应。本文概述的实验可以揭示与ad相关联的新alp基因。这些实验可以对ad发病机制中alp功能障碍的机制提供更深入的了解,并为将目前针对溶酶体贮积症的治疗策略重新用于ad的潜在治疗奠定基础。
[0313]
年龄是阿尔茨海默病(ad)的发展和进展的最大危险因素。在细胞水平上,衰老会降低自噬
‑
溶酶体途径(alp)的降解能力。尽管多项体外和体内研究表明,alp功能障碍导致
聚集的蛋白的缺陷清除有助于ad的发病机制,但尚不清楚alp功能的年龄依赖性和与ad相关联的下降背后的遗传变异。导致ad的基因和多个ad风险基因中罕见的功能变体会导致alp功能障碍。对分离的群体中单个alp基因的研究支持ad与溶酶体贮积症(lsd)之间的遗传重叠。然而,尚未完成对alp的各基因在普通群体中的遗传变异对发展ad风险的贡献及其在ad发病机制中的作用的系统和全面的评估。
[0314]
为了解决目前知识中的这一空白,本文描述了一些研究,以鉴定和优先考虑与发展ad风险相关联的alp的基因中具有较大效应大小的罕见功能变体。本文所述的耦合计算方法和实验数据的整合框架可以用于在体外和体内验证所选alp基因的功能效应。本文所述研究的可行性得到了无偏倚方法的支持,这些方法先前在有限数量的alp基因(pld3、grn、ctsf和sorl1)中发现了罕见变体,这些变体在家族性和散发性ad中均被过表达。因此,与在一般群体中发现的变异相比,对所有alp基因的分析预期揭示ad患者中罕见功能变体的富集。本文所述的研究克服了对分离的群体的研究中存在的分层群体导致的当前虚假关联,并在体外和体内的变体和基因水平上提供了互补的功能表征。
[0315]
(i)鉴定ad中富含罕见功能变体的alp基因
[0316]
假设alp基因中未经测试的罕见功能变体会影响发展ad的风险。为了鉴定这些风险变体,分析了来自33,350名对照的全外显子组测序(wes)数据[外显子组聚合联盟(exac)数据库,非芬兰欧洲人],以确定来自欧洲血统的个体的alp的每个基因中的基线遗传变异。接下来,通过将外显子组芯片和wes数据相结合,对2,000例ad病例和3,000名内部对照中的alp基因进行基于基因的分析。在独立样品中复制了这些结果,包括2,000例ad病例和2,000名对照,以及来自阿尔茨海默病测序项目(adsp)的10,000份公开可获得的wes数据样品。在折叠罕见变体后,已经鉴定出在至少12个溶酶体基因中具有较大效应大小(or>2.5)的预测的功能变体的富集(见结果)。
[0317]
(ii)确定alp的所选的候选基因对体外ad发病机制的功能作用
[0318]
推测与ad风险相关联的新型alp基因在体外ad发病机制中起作用。基于细胞的测定用于检查与ad风险相关联的候选alp基因中所选变体对酶活性、蛋白稳定性和/或mrna水平的影响及其对溶酶体功能的影响。检查了经验证的功能变体对淀粉样变性和aβ降解的影响。由dnajc5基因编码的cspα蛋白作为功能性溶酶体相关联蛋白的新作用已在之前被发现。此外,已收集了令人信服的数据,表明cspα在app加工和淀粉样变性中起作用(参见结果)。
[0319]
(iii)确定alp的所选的候选基因对体内ad病理的功能作用
[0320]
推测与ad风险相关联的新型alp基因在体内ad发病机制中起作用。在半合子或敲除(ko)naglu、npc1和dnajc5小鼠中,在由固有病理发展的时间定义的三个时间点,对ad病理的自发发展进行了定量和定性病理学调查。在良好表征的ad小鼠模型中测量了naglu、npc1和dnajc5基因的基因剂量对aβ斑块负荷的影响。
[0321]
本文所述的实验可以揭示与ad相关联的新alp基因。它们可以对ad发病机制中alp功能障碍的机制提供更深入的了解,这些知识是目前缺乏的并且有望产生新的治疗靶。本文所述的研究可以为将目前针对lsd的治疗策略重新用于ad的潜在治疗奠定基础。
[0322]
意义
[0323]
在人类基因组中存在至少430个与自噬
‑
溶酶体途径(alp)相关联的基因(38个自
噬基因、161个自噬调节基因、64个溶酶体基因和167个溶酶体调节基因)。所有alp基因中的38%(157个基因)的突变导致孟德尔病(omim),其中研究最多的是经典的溶酶体贮积症(lsd)。存在至少50种不同的lsd,其作为一个组,在每7,700例活产中出现频率为约1。lsd通常被视为儿科障碍,并且通常由完全功能丧失(lof)突变引起。然而,已经报道了携带亚形(hypomorphic)变体的成人发作形式的lsd。lsd为单基因障碍,但可以表现出复杂的临床特征。事实上,大约75%的lsd具有临床意义的神经系统组分。
[0324]
存在复杂疾病和孟德尔病的显著较高的共存率,表明在孟德尔障碍中受干扰的基因和途径也在相应的复杂疾病的病因中发挥作用。总体而言,近20%的与孟德尔表型有牵连的基因还含有或最接近负责复杂性状的全基因组关联研究(gwas)信号的变体。相比之下,所有基因中的约15%总体上属于孟德尔表型,这表明与孟德尔表型有牵连的基因富含gwas信号。近35%的alp基因组区域与gwas性状(gwas目录)相关联。事实上,alp途径中18.5%的基因在孟德尔病和常见病中发挥作用。大约22%是导致lsd的基因。尽管多条证据线(体外和体内)表明ad与lsd具有共同的分子机制,但尚不完全了解alp中与ad相关联的功能障碍背后的遗传变异。
[0325]
关于ad的病因的主要假设来自年龄相关和早发性疾病的遗传学研究,两者均与淀粉样β(aβ)肽的增加的产生和聚集有牵连。补充的假设表明,在没有孟德尔ad基因突变的患者中,包括aβ在内的致病蛋白的缺陷清除可能是ad的原因。在ad患者和ad小鼠模型中出现内体
‑
溶酶体和自噬调节障碍。在ad患者的脑中和在用溶酶体抑制剂治疗的小鼠中或在组织蛋白酶缺乏的小鼠中发现了自噬液泡(av)的大量神经元积累。溶酶体水解酶在ad患者的神经元中也被强烈地上调。此外,溶酶体在正常和异常的app加工及随后的淀粉样变性中也起重要作用。体外溶酶体功能的损害导致aβ产生的变化。暴露于氯化铵或巴非霉素a1会减少aβ分泌。用溶酶体蛋白酶抑制剂的治疗减少了溶酶体内淀粉样变性app片段的产生。在ad患者的脑脊液(csf)中溶酶体酶和溶酶体相关联蛋白水平发生改变。所有这些发现均支持以下假设:在ad中的alp内多个位点的积累“命中”会导致选择性失败,这削弱致病蛋白的清除。
[0326]
在选定的群体中,引起lsd的基因(诸如cstd、npc1和npc2)的罕见变体会增加ad的风险。此外,在引起lsd的基因缺陷的小鼠中进行的研究揭示了各基因在app加工和淀粉样蛋白生成中的独特作用。npc1、cln3和hexb基因缺陷的小鼠增加α
‑
ctf/β
‑
ctf和aβ40/42两者的水平。idua、sgsh、gba和tpp1基因缺陷的小鼠增加细胞内app/aβ水平,而无aβ斑块。与未检测到aβ42水平的对照相比,idua和sgsh基因缺陷的小鼠导致aβ40增加三倍。asah1和ppt1基因缺陷的小鼠减少细胞内app/aβ,而无斑块。然而,尚未完成对alp的各基因在一般群体中的遗传变异对发展ad的风险的贡献及其在ad发病机制中的作用的系统和全面的评估(参见,例如图5)。
[0327]
新生儿筛查研究显示,在健康人中报告的溶酶体酶活性水平存在十倍范围的差异。gba、npc1、galc、gaa、gla和idua基因中致病变体的杂合携带者表现出比对照显著更低的酶活性水平。此外,在尼曼
‑
匹克患者中,npc1中的致病变体的杂合携带者在受影响的主要途径下游表现出显著的代谢异常。到目前为止,似乎还没有聚焦于此类代谢改变和alp功能障碍的长期后果的系统研究。然而,少量的研究表明致病变体的杂合携带者具有神经退行性疾病的较高风险。
[0328]
本文描述的研究可以解决当前知识中的多个空白。首先,使用来自大型数据库的全外显子组测序(wes)数据以确定欧洲血统(ea)的个体的alp的每个基因中的实际基线遗传变异。其次,在ad病例中分析alp的每个基因中罕见功能变体的积累等位基因频率,以鉴定影响发展ad风险的候选基因。最后,在体外和体内验证了这些候选基因在ad的发病机制中的作用。通过鉴定ad中的alp基因中的特定缺陷,可以利用目前治疗lsd的多种治疗策略(包括基因疗法、酶替代、口服小分子底物减少疗法、小分子伴侣和自噬途径的药理学补救)进行ad的潜在治疗。
[0329]
创新
[0330]
本文概述的实验在其设计上是创新的,以定义与众所周知的与ad相关联的alp的功能障碍相关联的遗传变异,以及理解与ad相关联的alp基因中的罕见功能变体在体外和体内的作用。
[0331]
这些数据提供了令人信服的支持本文所述研究的可行性的证据。在包括naglu、npc1、ppt1、glb1和dnajc5基因(如下所述)在内的几个候选基因中,已经鉴定出所预测的罕见功能变体的富集。这些结果包括与ad风险相关联的新型alp基因,并为研究ad发病机制中alp功能障碍的机制开辟了新途径。例如,基于这一遗传分析,已经发现由dnajc5基因cspα编码的蛋白作为功能性溶酶体相关联蛋白的新的作用。此外,提供了令人信服的证据以表明,cspα在app加工和淀粉样蛋白生成中起作用。
[0332]
目前关于ad中alp基因的遗传变异的知识状态主要来自有限的研究,这些研究报告了复制率非常低的分离群体中的虚假关联。本文所述的研究可以克服该限制,因为该设计系统且全面地评估了在代表一般群体的非常大的样品中alp的每个基因中的遗传变异对发展ad的风险的贡献。因此,除了内部wes和外显子组芯片数据外,还使用了来自外显子组聚合联盟(exac)(n≈61,000)和阿尔茨海默病测序项目(adsp)(n≈10,000)的两个大型可公开获得的wes数据数据库中的数据,用于总的ad病例(n≈4000)和对照(n≈5000)。使用这些多个数据集,设计了一组分析,其将解决与ad相关联的alp中众所周知的功能障碍的遗传结构。
[0333]
存在alp的功能障碍与ad相关联的一般假设。然而,关于alp基因在ad发病机制中的作用尚无共识。这部分地是因为以下事实:大多数研究已经集中于使用可能不是适当细胞类型的经典表达系统(神经元样细胞系)的少数基因(例如,组织蛋白酶d)。此外,这些研究未评估适当的ad途径(app加工相对于tau聚集)。本文所述的研究并入了创新的综合框架,该框架将计算方法和实验数据相结合,以验证所选的alp基因在体外和体内的功能效应。基于细胞的测定补充了生化数据、活细胞测定、来自小鼠中特定脑细胞类型的rnaseq数据、人类ad病例和对照中的全基因组基因表达数据、来自不同阶段的人类ad病例的全基因组基因表达数据、来自不同年龄的人类的全基因组基因表达数据、来自与aβ斑块和神经原纤维缠结负荷相关的ad小鼠模型的全基因组基因表达数据。
[0334]
大多数采用alp基因的敲除小鼠的研究已经试图通过关注ad病理来了解其在ad发病机制中的作用。然而,由缺陷基因引起的病理是快速的,且与lsd更加相关联。这使对这些结果的解释复杂化,并且大多数时间都没有明确的理由。本文所述的研究可以基于由基于细胞的测定所支持的遗传分析的结果,评估来自所选alp基因的半合子小鼠中在不同时间点的ad病理(包括aβ斑块负荷)的易损脑区域。
[0335]
实验方法
[0336]
项目概述
[0337]
多项体外和体内研究表明,alp功能障碍有助于ad的发病机制。然而,尚不完全了解ad相关联的alp功能下降背后的遗传结构。为了解决这一问题,使用一种综合方法(其将大型数据集(内部数据库和公开可用的数据库两者)中对alp基因的预测的罕见功能变体的分析相结合)以优选考虑alp中的证明参与ad风险的候选基因(参见,例如第(i)节)。来自第(i)节中的分析的主要预期结果是基因的鉴定,而不是与ad相关联的单一变体的鉴定。接下来,在其相应的编码蛋白中验证所选变体的功能效应。在第(ii)节中,可以在基于细胞的测定中确定与ad风险相关联的选定基因的部分功能丧失的影响。该过程与来自人类疾病和小鼠模型的广泛数据挖掘过程相结合,以收集支持选定的候选alp基因在ad发病机制中的作用的生化、病理和细胞功能证据。在这个相互关联的过程中,计算数据定义并细化了实验数据,并且反之亦然(参见,例如第(ii)节)。最后,可以确定所选的alp基因是否与体内ad病理相关联(参见,例如第(iii)节)。这里概述的实验是基于遗传学和基于细胞的发现以及基于小鼠模型的可用性。此外,可以确定与ad相关联的选定alp基因的部分功能丧失是否会改变ad病理(易损脑区域中的aβ斑块)。检查对所选基因的完全缺失(
‑
/
‑
)和半合子(+/
‑
)的小鼠,以便区分ad病理是否是这些小鼠模型中的内在病理学的结果。此外,在老年半合子(+/
‑
)小鼠中检查了ad病理发展的选定基因。还在良好表征的ad小鼠模型中测量了选定alp基因的基因剂量对aβ斑块负荷的影响。来自这些实验的预期结果是单个alp基因对体内ad病理的贡献的确认。
[0338]
数据
[0339]
自噬
‑
溶酶体基因集(约430个基因)的列表已被手动编制,并且通过挖掘公共数据库(参见,例如生物信息学分析)和文献中的现有注释获得。alp功能障碍的最佳研究模型是lsd。lsd是由lof纯合的、复合杂合的突变或拷贝数变异(cnv)引起的一组遗传异质性孟德尔疾病。不同群体(主要是分离的群体)中的lsd的发生率非常低。因此,在一般群体中引起lsd的变体的预期频率极低。使用了exac数据库(其含有>60,000个测序的外显子),以在广泛的族裔抽样中估计引起lsd的变体的频率。只有三个x连锁引起lsd的基因(gla、ids和lamp2)具有功能丧失(lof)不耐受性(pli≥0.9)。在四十三个额外的引起lsd的基因中观察到的lof突变数低于在中性模型下的预期值。引起lsd的基因表现出广泛的蛋白质改变变体,从npc2基因中的三十二种到gaa基因中的423种。这些变体中有许多被预测为lof,可能在杂合状态下表现为亚形变体。这种大量的蛋白质改变变体可能解释了在人类中报道的溶酶体酶活性水平的广泛范围。有趣的是,在exac中,gaa基因表现出最高数目的蛋白改变变体和酶活性的较宽的可变性。
[0340]
大多数ad样品来自欧洲血统。因此,分析集中在非芬兰样品(约33,000个个体)中的引起lsd的基因(n=46)。在ncbi clinvar数据库中报告有约2740种引起lsd的变体。在exac样品中注释有288种引起lsd的变体:76%为错义变体,10%影响替代的剪接,并且12%为无义突变。大部分引起lsd的变体通过sift被预测为有害的(87%),并且通过polyphen2被预测为危险的(84%)。大多数(73%)引起lsd的变体位于高度保守的核苷酸中(grep评分>4)。这是对ea群体中引起lsd疾病的变体的频率的低估,因为cnv未包括在内。cnv是lsd的常见原因。此外,发现在exac中报告的几个lof变体,这些变体在ncbi clinvar数据库中未
被归类为引起lsd的变体。在每一个引起lsd的基因中都发现了引起lsd的变体,但是引起lsd的变体的数量与hyal1基因中发现的数量不同,与arsa基因中发现的二十个不同。每个基因的这些变体(杂合携带者的数量)的积累等位基因频率(cmaf)范围从cstd基因中的1.50e
‑
05
到npc2基因中的0.003。因此,应用1
×
10
‑3的cmaf阈值作为保守上界,因为在一般群体中比这更常见的变体预期不会是高度外显的引起lsd的变体。
[0341]
发现样品由来自523例不相关ad病例和386名对照的wes数据组成。表5显示了与ad相关联的顶级引起lsd的基因。
[0342]
表5.与内部对照和exac(非芬兰裔欧洲人)相比,在ad中引起lsd的基因中发现的罕见变体总结
[0343][0344][0345]
使用基于定义exac样品中引起lsd的变体的特征定义的纳入和排除标准对这些进行了分析。如所预期的,来自exac数据集(欧洲人,非芬兰血统)的基因特异性cmaf与来自内部ad数据库(欧洲血统的内部ad数据库)的cmaf高度一致(参见,例如图6)。每个引起lsd的基因(n=46)中符合纳入标准的变体按基因进行了核对:82%为错义变体,15%影响替代剪接,并且3%为无义突变。分析中包括的每个基因的变体的数量从arsb基因中的五个到npc1基因中的21个变化。将罕见蛋白改变变体(cmaf)的负荷与在对照和exac中观察到的负荷进行比较。对于这些基因中的大多数,与对照相比,在病例中存在过度变异,但仅发现与sgsh
基因(p=4.2
×
10
‑3;or=3.7,95%ci 1.4
‑
9.6)和cln8基因(p=1.0
×
10
‑2;or=8.9,95%ci 1.1
‑
68.1)的名义关联(参见,例如表5)。当与exac样品的cmaf比较时,十四个基因通过了非常严格的多重检验校正阈值p<1.0
×
10
‑4(0.05/450),并且十三个引起lsd的基因通过了基因水平显著性阈值p<2.4
×
10
‑6(0.05/20,000)(参见,例如表5)。接下来,使用两个另外的ad组群来复制表5中所列的发现,包括1722例ad病例,其中数据是使用人类外显子组芯片和来自1394例家族性ad(fad)病例的wes数据获得的(参见,例如表6)。
[0346]
表6.在两个另外的散发性ad(外显子组芯片)和家族性ad(fad)的样品中的复制发现。
[0347][0348]
如所预期的,在具有外显子组芯片数据的样品中仅复制了六个基因,但在fad的样品中复制了大部分关联(参见,例如表6)。这种不一致的最可能的解释是数据的覆盖深度。最好的实例是naglu基因中的结果,根据来自外显子组芯片的数据,没有变体符合纳入标准(参见,例如表6)。值得注意的是这样的事实:在复制样品中发现的关联在相同的方向并且效应大小相同。
[0349]
在三个样品中复制的alp基因之一是dnajc5基因,该dnajc5基因编码半胱氨酸串蛋白α(cspα),并且其突变导致成人发作lsd。cspα被定位于神经突中的质膜。其在神经元样细胞类型(n2a)中具有弥漫性胞质定位,而且还有一部分内源性cspα与lamp2被共同定位于胞体中(参见,例如图7a)。亚细胞分级显示,相当比例的cspα与另一种溶酶体标记物(lamp1)共沉积(参见,例如图7b)。这些结果表明,内源性cspα是一种溶酶体相关联蛋白。如所预期的,与空载体相比,在表达引起lsd的突变(p.l115r)的细胞中存在显著更高水平的溶酶体跟踪信号。相比之下,与表达cspα
‑
p.l115r或空载体的细胞相比,经cspα
‑
wt转导的细胞显著降低了溶酶体跟踪信号(参见,例如图7c),这表明cspα可能参与溶酶体ph的调节。与空载体相比,引起lsd的突变(p.l115r)的表达导致细胞内和分泌的溶酶体酶的显著升高(参见,例如图7d和图7e)。相反,与用空载体或cspα
‑
p.l115r转导的细胞相比,cspα
‑
wt的过表达导致细胞内和分泌的溶酶体酶的显著减少(参见,例如图7d和图7e),表明溶酶体转运和胞吐受到cspα的影响。
[0350]
dnajc5转录物在神经元中和在最易受ad病理影响的脑区域中被高度表达。在来自
青年(<40岁)、中年(40
‑
70岁)和正常老年(70
‑
94岁)的额叶皮质区的神经病理学正常脑样品中发现dnajc5转录水平随年龄的降低(p=0.0003;geo数据库;gds5204系列)(参见,例如图8a)。在ad和对照的激光捕获微解剖的非缠结神经元中,与年龄匹配的对照相比,在ad病例中的dnajc5转录水平显著较低(p=<0.0001,参见,例如图8b,左图)(geo数据库;gse5281系列)。在不同的研究中重复了这一发现(geo数据库;gse15222系列)(参见,例如图8b,右图)。还发现,与野生型小鼠中的水平(图8c中的黑线)相比,在两种ad小鼠模型(tau,p.p301l;图8c中的蓝线)和(app,p.k670n/p.m671l;图8c中的红线)的皮质中,dnajc5转录水平表现出年龄依赖性降低,并且与ad病理的发展成反比(右图,参见,例如图8c)。所有这些结果表明,cspα可能参与了ad的发病机制。
[0351]
在cspα中具有引起lsd的变体(p.l115r)的患者的大脑未呈现出aβ斑块或神经原纤维缠结。然而,组织学分析揭示,皮质神经元(人lsd,参见,例如图9a)中app/aβ(抗体4g8)的显著的细胞内积累。因此,在体外测试了cspα在淀粉样蛋白生成中的作用。在n2a695细胞中app/aβ免疫反应性与溶酶体标记物共定位(空载体,参见,例如图9b)。敲低cspα在n2a695细胞中的表达降低了aβ/app水平及其与lamp
‑
1的共定位(shrna,参见,例如图9b)。在cspα中稳定地表达引起lsd的变体的n2a695细胞表现出app/aβ的细胞内积累(p.l115r,参见,例如图9b)。表达hcspα
‑
p.l115r的n2a695细胞释放的aβ40和aβ42水平显著高于用空载体转导至培养基的细胞(参见,例如图9c,左图)。相反,表达特异性shrna
‑
cspα的n2a695细胞分泌显著更少的细胞外aβ40和aβ42水平(参见,例如图9c,左图)。当与用空载体转导时相比,表达hcspα
‑
p.l115r的n2a695细胞在细胞内积累了更多的aβ40(参见,例如图9c,右图)。在不同组之间的aβ42水平没有差异(参见,例如图9c,右图)。用shrna
‑
cspα转导的细胞显示全长app、α
‑
ctf/β
‑
ctf、cspα和sappα的水平降低(参见,例如图9d,图)。在hcspα
‑
p.l115r中显示全长app和α
‑
ctf/β
‑
ctf的增加,而sappα的水平没有变化(参见,例如图9d,图)。这些结果表明,cspα在淀粉样蛋白生成中以及可能在ad发病机制中的新的且意外的作用已被发现。
[0352]
研究设计与方法
[0353]
(i)鉴定ad中富含罕见功能变体的alp基因
[0354]
假设alp的基因中未经测试的罕见功能变体会影响发展ad的风险。如本文所述,在大数据集(内部数据库和公开可用的数据库两者)中结合alp的基因中预测的罕见功能变体的分析的综合方法可以用于优先考虑alp中有证据表明参与ad风险的候选基因。
[0355]
定义罕见变异的等位基因频率阈值
[0356]
使用从引起lsd的变体的分析中获得的信息(包括引起lsd的变体的最高maf、sift、polyphen2或gerp评分),定义了以下纳入标准:1)ad病例中的调用率>98%,2)每个变体的maf<0.01%,3)由exac或系综注释为错义的指定规范转录物中的仅可能的蛋白改变变体,4)框架移位,5)无义,6)影响剪接供体和受体区域的变体,和7)如果在ncbi clinvar数据库中有致病性证据并且位于3'或5'utr区域。在exac中未发现的变体,或ncbi clinvar数据库中不存在的同义、内含子、3'或5'utr变体或maf>0.01%、exac和clinvar中的错误调用被排除在外。为了确保群体特异性变体对本分析没有混杂效应,基于主组分结果选择了个体。
[0357]
研究群体
[0358]
已经从2,000名个体中获得了wes。来自knight
‑
adrc的总共3,000名个体可以访问
外显子组芯片数据。也可以获得adni和knight
‑
adrc样品的干净和估算的gwas数据。存在可用于阿尔茨海默病神经影像学倡议的dna(adni;600例病例和200名对照)、nia
‑
load(867例不相关病例和645例不相关对照)、knight阿尔茨海默病研究中心(knight
‑
adrc;779例病例,555名对照)和西班牙数据集(167例病例和534名对照)。之前已发布了这些数据集的描述。使用与国家神经病学和交流障碍和中风研究所
‑
阿尔茨海默病和相关障碍协会对可能的ad(national institute of neurological and communication disorders and stroke
‑
alzheimer's disease and related disorders association for probable ad)等效的标准,每个病例都均接受了阿尔茨海默型痴呆的诊断。对照组接受与病例相同的评估,但认知正常。所有个体均为欧洲裔,并从所有参与者获得了书面同意。数据下载自exac(版本0.3.1,2015年3月)。在分析中仅包括具有覆盖至>30
×
的中位序列深度的高比例的编码区以及仅高质量(带通滤光器)变体的基因。数据从adsp下载。发现阶段数据集含有来自113个家庭的584名受试者的wgs数据,以及来自受ad多重影响的家庭的853名(682例病例[510名非西班牙裔,172名西班牙裔])和171名西班牙裔对照受试者的系谱数据。
[0359]
全外显子组测序数据
[0360]
存在来自2,000名个体的wes。使用sureselect 52mb靶富集系统(agilent)进行外显子组富集。dna通过配对末端阅读(illumina hiseq2000)来测序。使用novoalign和sam工具执行对比和变体调用。这些方法以前使用过,对基因型调用具有良好的特异性和敏感性。gwas数据用于序列调用的质量控制。在这些研究中,发现在外显子组测序调用与gwas数据之间符合率>98%。
[0361]
人外显子组芯片数据
[0362]
访问来自knight
‑
adrc的总共3,000名个体的外显子组芯片数据。illumina和affymetrix已经开发了廉价的现成基因分型芯片,其被称为“外显子组芯片”,其含有外显子内的变体,这些变体已在外显子组变体服务器数据库中被报告至少两次。外显子组芯片中包括的大多数编码变体都是非常罕见的变体,maf<0.01。这些阵列提供了一种快速且简单的方法以分析低频变体。然而,这些阵列并不包括所有的编码变体。
[0363]
外显子组芯片质量控制(qc)
[0364]
使用其他地方描述的用于调用illumina外显子组芯片数据的最佳实践进行基因分型调用。用于外显子组芯片的qc与用于gwas的qc步骤相似,但由于低的maf,未去除变体。提供了外显子组芯片的原始数据,并且在单个变体或基因水平上检查了聚类的任何显著关联。
[0365]
负荷测试
[0366]
为了适应具有适中效应大小的罕见变体的效应,使用了已开发的经验证的统计方法来分析与罕见变体的关联。简而言之,基于基因的方法将区域内的罕见变体折叠成单个值,并且然后测试区域内的罕见变体与感兴趣性状之间的关联。序列核关联检验(skat)用于检验基因区域内的状态与罕见变体之间的关联。与其他基于基因的方法相比,skat的优势在于,skat可以解释同一基因中在不同方向上具有影响的变体,并对混杂的协变量进行调整。计算替代等位基因与最常见等位基因相比的具有95%置信区间的优势比。如果在等位基因检验中检测到关联,则另外的分析确定加性模型或显性模型是否更适合。独立的病例对照样品被用于复制来自发现数据集的发现。对每个不同数据集的分析是分别进行的。
进行联合分析以合并p值和or。该方法已被成功地用于先前的研究中,以鉴定ad的新基因。
[0367]
gwas数据
[0368]
之前已对大多数(>90%)样品进行了全基因组基因分型。采用多种阵列进行基因分型,包括neurox
‑
chip(wu和ppmi)。在关联分析或估算之前,所有样品和基因型都经历严格的质量控制(qc)。通过应用snp和个体的最小调用率(98%)和最小次要等位基因频率(maf=0.02),来清理基因型数据。不属于哈迪
‑
温伯格(hardy
‑
weinberg)平衡(p<1
×
10
‑6)的snp被排除。使用成对全基因组范围的按血统同一性比例的估计来进行未预料到的重复和神秘相关度的测试。
[0369]
估算(imputation)
[0370]
1,000个基因组项目数据(第3阶段,2014年11月发布)和impute2软件被用于估算多达600万个snp。移除r2<0.5、次要等位基因频率(maf)<0.02、超出哈迪
‑
温伯格平衡(p<1
×
10
‑6)、调用率<95%或gprobs评分<0.90的snp。在先前的gwas和估算过程中,总共6,815,690个snp通过了qc过程。
[0371]
群体结构
[0372]
鉴于gwas数据的可用性,在样品上使用eigenstrat以及hapmap样品作为锚,以确认自我报告的种族/民族。来自群体分层分析的前三个主成分因子(pc)作为协变量被包含在分析中。
[0373]
数据存储和管理
[0374]
对于每个外显子组产生高达150gb的处理的数据,并且需要三天来比对序列和执行snp调用。因此,有必要具有大型且安全的数据存储系统。所有生成的数据都保存在具有3太字节(tb)空间的linux服务器上。一旦数据被处理,并且通过外显子组测序检测到的序列变体通过基因分型被确认,就应用了已开发并用于其他外显子组测序项目的高效和有效的用于数据管理、质量控制、清洁、注释和分析的方法。
[0375]
生物信息学分析
[0376]
以下公开可用的数据库被用于补充分析:在线人类孟德尔遗传(online mendelian inheritance in man)(omim)、外显子组变体服务器(exome variant server)exac、gwas目录、gerp4、clinvar数据库和人类自噬数据库。
[0377]
功效分析
[0378]
为了确定检测与发作年龄相关联的遗传变体的功效,在sas中使用proc power运行分析。使用在0.05至0.50范围内的次要等位基因频率、1.2至3.6的or以及4,000至7,000个样品量运行分析。对于单变体分析,α被调整为5
×
10
‑8;并且对于基于基因的分析,α被调整为5
×
10
‑6。基于这些结果,存在大约80%的功效以检测or>1.19的效果(或<0.84)。
[0379]
预期的结果
[0380]
本文所述研究的可行性得到了先前无偏倚方法的支持,这些方法已经发现在家族性和散发性ad中过表达的有限数量的alp基因(pld3、grn、ctsf和sorl1)中的罕见变体。(i)的目的是从人类基因组中属于alp的约430个基因中鉴定与ad风险相关联的基因。聚焦于引起lsd的基因(n=46)的研究表明,在与ad风险相关联且效应大小较大的alp中的至少12个额外的新基因可以被鉴定(参见,例如表5),并且这些基因可以在覆盖深度适当的额外样品中被复制(参见,例如表6)。这些结果支持了以下假设:与在一般群体中发现的变异相比,在
ad患者中alp基因的变异更高。因此,预期的是将揭示ad风险与分析中包括的剩余约384个alp基因的数目的新的关联。
[0381]
结果和功效计算表明,在基于基因的分析中,存在足够的功效以检测>2.7的平均优势比。如果alp基因的关联未能与病例对照设计复制,则可以使用内表型设计。因此,可以通过对lsd基因和以下每种csf生物标记物(t
‑
tau、p
‑
tau和aβ42)进行单变体和基于基因的分析,来确定alp基因中的变体对ad的csf生物标记物水平的影响。为了评估alp基因的调控基因组区域是否可能参与发展ad的风险,可以分析在来自由国际阿尔茨海默病基因组学项目(international genomics of alzheimer’s project)(i
‑
gap)先前发表的gwas的数据中alp基因的关联,该数据由总共25,580例ad病例和48,466名对照组成。一种补充方法可以是检查alp基因是否影响发作年龄(aao),因此,可以检查来自之前发表的gwas的研究与ad发作年龄相关联的遗传变体的数据。
[0382]
使用全外显子组测序,预计可以鉴定出大多数蛋白改变变体。然而,很明显,用rnaseq数据补充wes数据可以鉴定更多潜在的功能变体,尤其是影响剪接供体和受体区域的变体。目前正在从已获得wes数据的ad病例和对照的500个脑组织样品中生成rnaseq数据,其允许分析影响ad发病机制中alp基因中的剪接的变体的影响。还正在进行100个个体的全基因组测序,以对基因组的非编码基因组区域的作用进行更详细的分析。
[0383]
(ii)确定alp的选定的候选基因对体内ad发病机制的功能作用
[0384]
推测与ad风险相关联的新型alp基因在体外ad发病机制中起作用。如本文所述,基于细胞的测定可以用于检查与ad风险相关联的候选alp基因中所选变体对酶活性、蛋白稳定性和/或mrna水平的影响及其对溶酶体功能的影响。经验证的功能变体对淀粉样蛋白生成、app加工和aβ降解的影响可以如下文所述进行检查。
[0385]
方法和分析
[0386]
评估在(i)中鉴定的与ad相关联的每种alp基因的所有变体超出了本文所述研究的范围。因此,在(i)中鉴定出的符合以下标准的基因中的前3
‑
5个变体被优先考虑。1)存在可用(商业上或通过合作者)的小鼠模型,其可以用于获得原代神经元/小胶质细胞并进行(iii)中概述的体内实验。2)存在可用的生物化学和/或基于细胞的测定以用于检测其功能的变化。3)在圣路易斯华盛顿大学(washington university in saint louis)有具备足够专业知识的合作者来协助数据的分析。因此,考虑到来自发现和复制样品的数据强度以及所有上述标准,已经选择了这三个基因naglu、npc1和dnajc5。专业知识和适当的协作可以用于执行本文所述的所有实验。
[0387]
克隆选定的alp基因
[0388]
表征野生型和突变候选基因对蛋白表达和正常功能的影响非常重要。cdna克隆购自origene或英杰公司的候选基因。使用定点诱变试剂盒(quikchange ii(安捷伦科技(agilent technology),美国加利福尼亚州圣克拉拉(santa clara,ca,usa))对选定的变体进行工程改造。
[0389]
慢病毒产生
[0390]
野生型和突变型cdna被亚克隆到plenti
‑
iii
‑
pgk载体(应用生物材料公司(applied biological materials inc),加拿大里士满(richmond,canada))中,该载体携带嘌呤霉素抗性基因。如前所述,将所得的慢病毒载体与编码vsv
‑
g、gag
‑
pol和rev的质粒
一起共转染到hek
‑
293t包装细胞中。根据先前公布的方案来收集病毒上清液。用未浓缩的病毒上清液培养n2a695细胞持续24小时,并用5μg/ml的嘌呤霉素选择细胞持续四周。使用携带对naglu、npc1和dnajc5基因特异性的shrna的慢病毒载体的敲低模型也在n2a695细胞中产生。
[0391]
基于细胞的测定
[0392]
使用以下细胞系:人胚胎肾(hek293
‑
t)、n2a和n2a695(稳定地表达人app695wt(被称为n2a695)的小鼠神经母细胞瘤细胞,其常规用于研究app加工)(参见,例如图7a)。来自naglu、npc1和dnajc5半合子或敲低小鼠模型的原代神经元或小胶质细胞也用(i)中鉴定的变体转导。如前所述进行原代神经元培养。
[0393]
mrna和蛋白水平的影响
[0394]
使用特异性引物的定量实时pcr被用于检测所选的变体对mrna水平和剪接的影响。使用以下抗cspα(adi
‑
vap
‑
sv003
‑
e,恩佐生命科学(enzo life sciences))抗naglu(ab137685,abcam)和抗npc1抗体进行蛋白免疫印迹。如前所述进行naglu活性的荧光测定(参见,例如图7d和图7e)。简而言之,在日立f
‑
2000荧光分光光度计(日立(hitachi),加利福尼亚州普莱森顿(pleasanton,ca))中,使用范围为0.02至5mm的4
‑
甲基伞形酮(sigma公司,密苏里州圣路易斯)的标准曲线,在448nm发射和365nm激发下测量4
‑
甲基伞形酮
‑
n
‑
乙酰基
‑
α
‑
氨基葡萄糖苷裂解。npc
‑
1特异性测定如先前所公布的进行。
[0395]
溶酶体功能
[0396]
使用溶酶体跟踪器,以使用流式细胞术来定量每个细胞的酸性隔室的数目(参见,例如图7c)。溶酶体ph由溶酶体传感器黄色/蓝色(lysosensor yellow/blue)葡聚糖(dnd
‑
160)测量。通过吖啶橙来监测溶酶体膜的完整性。使用组织和培养细胞的溶酶体富集试剂盒(赛默飞世尔科技公司(thermo scientific))进行亚细胞分级分离,使用lamp1、rab7和eea1作为溶酶体、晚期和早期内体标记物的对照(参见,例如图7b)。通过免疫荧光共聚焦图像对该测定进行补充,以在溶酶体(lamp
‑
1或lamp
‑
2)、早期内体(eea1)、晚期内体(rab7)、质膜(flotillin)标记物内共定位所选的蛋白(参见,例如图7a)。
[0397]
基于细胞裂解物的溶酶体酶活性
[0398]
通过ppt
‑
1、β
‑
gluc和β
‑
hexa的荧光测定法进行溶酶体酶活性的细胞内和细胞外二级升高(参见,例如图7d和图7e)。
[0399]
巨噬细胞的药理学调节
[0400]
用自噬系统的激活剂和抑制剂(雷帕霉素、torin1、torin 2、甲胺、布雷非德菌素a和spautin
‑
1)以及伴侣介导的自噬(ar7)处理转导的细胞。lc3和p62的蛋白免疫印迹可以被用作巨噬细胞活化的间接指标。溶酶体营养剂诸如氯喹、氯化铵(nh4cl)和亮抑蛋白酶肽被用作阳性对照。
[0401]
对app加工和周转的影响
[0402]
用蛋白合成抑制剂环己酰亚胺来处理n2a695细胞,以定量app半衰期。分别通过蛋白免疫印迹和rt
‑
qpcr来测量app加工机制(psen1、呆蛋白、adam10、adam17和bace1)的蛋白和转录水平。为了测量所选alp基因对细胞表面app的动态影响,在4℃下使用非膜通透性切割生物素衍生物(磺基
‑
nhs
‑
ss
‑
生物素)来标记质膜上的app。如前所述,通过链霉亲和素ip和app免疫印迹法来测量细胞表面app。
[0403]
aβ40/aβ42水平
[0404]
如前所述,通过夹心elisa来检测来自转导的n2a695的细胞裂解物和细胞培养基中的aβ物种(参见,例如图9c)。用小鼠单克隆包被抗体hj2(抗aβ35
‑
40)和hj7.4(抗aβ37
‑
42)捕获aβx
‑
40肽和aβx
‑
42肽。hj5.1(抗aβ13
‑
28)(一种靶向中心结构域的生物素化抗体)或hj3.5(抗aβ1
‑
13)(其靶向n
‑
末端氨基酸)被用作检测抗体,然后是链霉亲和素
‑
聚
‑
hrp
‑
40(菲茨杰拉德工业公司(fitzgerald industries))。
[0405]
app的α/β分泌酶加工
[0406]
通过蛋白免疫印迹来测量细胞上清液和细胞内片段(α
‑
ctf和β
‑
ctf)中的app裂解产物(sappβ和sappα)。以下抗体22c11和ct695被用于表征全长以及α
‑
ctf和β
‑
ctf片段(参见,例如图9d)。
[0407]
aβ摄取/降解
[0408]
用选定的变体或特异性shrna转导的原代小胶质细胞用250nm的合成aβ42处理2小时。为了测量aβ的摄取,对aβ42处理的细胞进行洗涤和胰蛋白酶化以去除表面结合的aβ,溶解,并且通过夹心elisa测量细胞内aβ42(参见,例如图9c)。通过改变细胞被暴露于aβ42的时间(0、5、10、30分钟和1、2、4、8、12、24小时)来测量aβ摄取率。为了测量aβ降解,用250nm的合成aβ42处理细胞2小时,彻底洗涤,并且在新鲜培养基中孵育。然后对细胞进行洗涤、胰蛋白酶解、裂解,并且在0、2、4、8、12、24小时后测量细胞内aβ42。细胞内aβ半衰期的计算假定为一级动力学(如先前所公布的)。
[0409]
生物信息学分析
[0410]
下列公开可用的数据库被用于补充分析:基因表达综合数据库(gene expression omnibus)、脑rna
‑
seq2、来自小鼠痴呆网络(小鼠demnet)、polyphen2、sift、人类剪接查找器和小鼠基因组信息学的基因表达数据。
[0411]
预期的结果
[0412]
基于这里描述的用于检测所预测的罕见功能变体的具体设计以及dnajc5的确证结果,预计(i)中鉴定的alp基因风险变体会导致部分功能丧失并改变溶酶体功能。预计在这些蛋白的功能中将检测到5
‑
20%的残留活性。还预计alp基因的过表达、下调或剪接改变变体会影响app代谢和淀粉样蛋白生成。可以使用计算资源和来自文献的挖掘数据来仔细地选择细胞类型和功能测定,包括每个alp基因的aβ或tau代谢测定。此外,预期本文描述的实验允许评估alp基因中的选定变体对神经元和小胶质细胞存活的影响。
[0413]
如果在naglu、npc1和dnajc5基因中未观察到wt与风险变体之间的显著差异,则这可能是由于过表达掩盖了功能的微妙变化。因此,ad风险变体会引起微小变化,这种变化可能会在一生中的疾病中表现出来,但在细胞培养的几天内观察效果可能是一种挑战。因此,app突变可能被过表达,或者alp功能可能在药理学上发生改变。来自ad小鼠模型(诸如5xfad转基因小鼠(34840
‑
jax))的原代神经元可以用于naglu、npc1和dnajc5基因中的选定变体并用该选定变体转导,并测试了对app加工和tau聚集的影响。可以使用来自携带naglu、npc1和dnajc5基因中的变体的ad患者的ipsc衍生神经元,并且可以将此类变体对app代谢的影响与对照进行比较。基因组编辑方法(诸如crispr技术)可以用于在alp基因中引入所选的变体并执行上述功能测定。可以用亚致死剂量的自噬抑制剂的溶酶体营养剂来处理在naglu、npc1和dnajc5基因中的稳定地表达选定变体的n2a695细胞或原代神经元或
神经胶质细胞,并测量对app加工的影响。
[0414]
在体外鉴定naglu、npc1和dnajc5中影响ad的风险和发病机制的变体后,接下来的步骤是利用lsd疗法的进展。利用诸如酶替代、底物减少、分子伴侣或alp的药理学调节的治疗策略,并在体外测试其对ad发病机制测定的影响。
[0415]
(iii)确定alp的所选候选基因在体内对ad病理的功能作用
[0416]
假定与ad风险相关的新型alp基因在体内ad发病机制中起作用。如本文所述,可以在半合子或敲除(ko)naglu、npc1和dnajc5小鼠在由内在病理的发展时间所定义的三个时间点对ad病理的自发发展进行定量和定性病理学调查。还可以在良好表征的早发作的家族性ad、5xfad转基因小鼠模型(34840
‑
jax)中测量naglu、npc1和dnajc5基因的基因剂量对aβ斑块负荷的影响。
[0417]
方法和分析
[0418]
评估(i)中鉴定的与ad相关联的每个alp基因超出了本文所述实验的范围。因此,已经定义了选择用于后续体内研究的小鼠模型的选择的纳入标准:其必须可获得(在商业上或通过合作者),必须存在功能性测定、稳健的脑病理、稳健的行为改变(理想情况下为短寿命)以及对淀粉样蛋白生成的影响的体外验证。
[0419]
小鼠组
[0420]
为该实验产生3组(18只小鼠/组)naglu、npc1和dnajc5小鼠:1)正常同窝仔畜,2)半合子(+/
‑
)小鼠,和3)在三个不同时间点的缺陷(
‑
/
‑
)小鼠。naglu ko小鼠的中位生存期为12个月。然而,脑病理早在3个月时是明显的。因此,在2个月、4个月和8个月时在naglu小鼠中检查ad病理。dnajc5 ko小鼠的中位生存期为60天,并且脑病理早在30天是明显的。因此,在第21天、第30天和第40天检查ad病理。npc1 ko小鼠的中位生存期为75天,因此在第30、50和70天在npc1 ko小鼠中检查ad病理。
[0421]
也使用了ad小鼠模型5xfad。5xfad小鼠在六周龄时积累神经内aβ
‑
42。在老年小鼠中,海马中的淀粉样蛋白沉积出现在第二个月,并在整个大脑中传播。为了进一步确定naglu、npc1和dnajc5缺乏是否会加剧现有的淀粉样蛋白生成过程,将naglu、npc1和cspα小鼠与5xfad转基因小鼠杂交。为该实验产生3组(27只小鼠/组)。基因剂量对aβ斑块负荷的影响是通过在5xfad/naglu(+/
‑
)、5xfad/npc
‑
1(+/
‑
)和5xfad/dnajc5(+/
‑
)小鼠中测量三周时aβ
‑
42脑水平、1个月和两个月时淀粉样蛋白沉积来确定的。为该实验产生3组(27只小鼠/组)。通过在针对评估每只ko小鼠的内在病理而描述的相同时间点分析5xfad/naglu(
‑
/
‑
)、5xfad/npc
‑
1(
‑
/
‑
)和5xfad/dnajc5(
‑
/
‑
)小鼠,来评估所选基因的完全缺失对5xfad病理的影响。在组织学分析期间,实验者对动物的基因型和年龄视而不见。每只小鼠都被分配随机的id号。
[0422]
淀粉样斑块定量
[0423]
每隔300μm从嘴侧前连合到尾侧海马来采集五十个微米厚的振动体脑切片。对于斑块成像,切片用thios染色或用hj3.4抗aβ抗体免疫染色。使用nanozoomer数字扫描仪(滨松光电公司(hamamatsu photonics))获得染色的脑切片的高分辨率数字图像。使用nih imagej测量海马或梨状皮质区域中的斑块覆盖的总面积,并以每张切片的总面积百分比表示。对来自n=4个切片的结果进行平均,以代表每只动物。
[0424]
aβ40/aβ42水平
[0425]
为了检测青年小鼠的海马中的总aβ,将解剖的组织依次在pbs中匀浆,然后在未观察到斑块的年龄时在ripa缓冲液中匀浆,以获得可溶于去污剂的aβ,并且混合样品用于分析。在老年小鼠(当斑块丰富时)中,海马组织依次在pbs中匀浆,然后在tbs(ph8.0)中的5m胍中匀浆(以提取原纤维性和膜结合的aβ)。aβ40/aβ42水平采用elisa进行定量,如(ii)所述。
[0426]
预期的结果
[0427]
基于遗传分析报告的较大效应大小,预期在半合子的和缺陷已被发现与ad风险相关联并经体外测定验证的候选alp基因的小鼠中,发现的ad病理多于相应的年龄匹配对照中的ad病理。也期望看到对ad病理的基因剂量效应;半合子和ko小鼠naglu、npc1和dnajc5基因会加速和恶化5xfad小鼠中的aβ斑块负荷的负荷。
[0428]
如果在半合子和缺陷小鼠的脑中未发现ad病理,则生成携带(ii)中验证的最显著变体的aav2/9载体,并立体定向注射到半合子小鼠的海马中,并重新评估ad病理的存在。如果发现ad病理,也可以通过注射携带缺陷基因的野生型拷贝的aav2/9载体并重新检测ad病理来尝试挽救实验。另一种测试naglu、npc1和dnajc5基因在ad病理中的作用的方法是,注射携带经验证的shrna/rnai的aav2/9载体,以在5xfad转基因小鼠中敲低该基因,并测试是否存在对ad病理的影响。
[0429]
可以在体内测试lsd的疗法(诸如酶替代、底物减少、分子伴侣和alp的药理学调节)对ad发病机制测定的影响。结合来自本文所述研究的结果以及naglu活性的荧光测定法和npc1生物标记物的质谱测定法的可用性,可以筛选大组群ad病例和对照的csf、血浆、血清或干血点,以检测可以用作ad的生物标记物的特定缺陷。为了确定naglu、npc1和dnajc5缺乏是否会加剧tau病理,将naglu、npc1和dnajc5小鼠与表达tau,p.p301l(015815
‑
jax)的小鼠杂交。到4个月大时,tau,p.p301l小鼠在皮质中出现缠结。因此,在与naglu、npc1和dnajc5小鼠杂交的小鼠中,在一个月大时测量tau水平。已经优化了测量小鼠脑中高磷酸化的tau负荷的条件。通过elisa定量tau脑水平以前也已经被优化。使用此类crispr技术的基因组编辑方法,以生成alp基因中的变体的敲入小鼠,其对体外测定具有最强的影响。这些敲入小鼠还与包括5xfad在内的ad小鼠模型和表达tau,p.p301l的小鼠杂交。
[0430]
实例3:鉴定阿尔茨海默病(ad)中导致自噬
‑
溶酶体途径(alp)的功能障碍的遗传变异
[0431]
本实例描述了自噬
‑
溶酶体途径的基因中的罕见功能变体在阿尔茨海默病中的作用。
[0432]
尽管多项体外和体内研究表明自噬溶酶体途径(alp)功能障碍有助于ad的发病机制,但尚未完全了解alp功能的年龄依赖性和与ad相关联的下降背后的遗传变异。引起ad的基因和多个ad风险基因中罕见的功能变体会导致alp功能障碍。然而,尚未完成对alp中每个基因的一般群体内的遗传变异对发展ad的风险的贡献及其在ad发病机制中的作用的系统和全面的评估。为了解决现有知识中的这一空白,本文描述了一种强有力的方法,以鉴定并优先考虑alp的基因中与发展ad的风险相关联的罕见功能性杂合变体。结合计算方法和实验数据的创新性整合框架可以用于验证所选的alp基因在体外和体内的功能效应。首先,将来自33,350名非芬兰籍欧洲对照的alp的各基因中罕见功能变体的负荷与2,000例ad病例和3,000名对照进行比较。在额外的独立样品中复制了这些结果,包括2,000例ad病例和
2,000名对照以及来自阿尔茨海默病测序项目(adsp)的10,000个公开可用的样品。其次,使用生物化学和基于细胞的测定来全面表征与ad相关联的候选alp蛋白中所选遗传变体的功能作用。研究了突变的蛋白对alp功能的影响以及对全长app水平、app转运、神经元中的aβ生成和通过胶质细胞的aβ降解的影响。最后,可以确定naglu、npc1和dnajc5基因的单倍体不足性是否会加速良好表征的ad小鼠模型中存在的ad病理。对遗传性轻度慢性溶酶体损伤对涉及aβ的ad相关表型的影响进行了定量病理学调查。本文所述的实验可以揭示与ad相关联的新alp基因。它们可以为ad发病机制中alp功能障碍的机制提供更深入的了解。
[0433]
本文所述研究的目的是鉴定阿尔茨海默病(ad)发病机制中涉及的自噬
‑
溶酶体途径(alp)的功能障碍背后的遗传变异。本文所述的研究并入了创新的综合框架,该框架将计算方法和实验数据相结合,以验证所选的alp基因在体外和体内的功能效应。本文概述的实验可以揭示与ad相关联的新alp基因。它们可以为ad发病机制中alp功能障碍的机制提供更深入的了解。
[0434]
自噬
‑
溶酶体途径的基因中的罕见功能变体在阿尔茨海默病中的作用
[0435]
年龄是阿尔茨海默病(ad)的发展和进展的最大危险因素。衰老也会降低自噬
‑
溶酶体途径(alp)的降解能力。尽管多项体外和体内研究表明alp功能障碍有助于ad的发病机制,但尚不完全了解alp功能的年龄依赖性和ad相关联下降背后的遗传变异。主要假设是,轻度形式的遗传性alp功能障碍(其由额外的年龄相关alp损伤加重)会导致ad病理的发展。因此,神经元中的遗传性alp损伤可以增加淀粉样蛋白的生成,而神经胶质细胞中的alp损伤可以降低其降解淀粉样斑块的能力。因此,产生与清除之间的平衡决定了aβ水平和形成淀粉样斑块的倾向。
[0436]
聚焦于分离的群体中少数alp基因(例如组织蛋白酶d)的单一编码变体的小样品量研究以及来自大规模全基因组关联研究(large genome
‑
wide association studies)(gwas)(例如,sqstm1)的内含子“命中”支持alp基因中的遗传变体在ad风险中的作用。此外,引起ad的基因(例如早老素)的罕见突变和ad风险基因(例如sorl1)的功能性编码变体会导致alp功能障碍。人类基因组编码至少430个与alp相关联的基因。然而,尚未完成对alp的各基因中编码变体对发展ad的风险的贡献的系统和全面的评估。如本文所述,第(i)节可以解决这一空白。
[0437]
将全外显子组测序(wes)数据与大量ad病例和对照数据库相结合,先前发现推定alp基因(磷脂酶d家族,成员3,pld3)的罕见变体与ad风险相关联。pld3是一种转录因子eb(tfeb)应答基因,并且似乎通过溶酶体介导的机制影响淀粉样前体蛋白(app)加工。有数据表明,在晚发型散发性ad中,预测的功能性杂合变体在几个额外的溶酶体基因中富集。为了验证那些关联,发现突触伴侣半胱氨酸串蛋白(cspα)作为功能性溶酶体相关联蛋白的互补作用。此外,已收集的数据显示,在ad患者和小鼠模型的大脑中cspα转录水平降低。此外,cspα的突变会影响体外的自噬体/溶酶体融合并且影响体内aβ生成。在这些分析中,编码细胞内胆固醇转运体1(npc1)和n
‑
乙酰基
‑
α
‑
氨基葡萄糖苷酶(naglu)的基因也与ad风险相关联。此外,在正常人脑样品中,发现npc1和naglu转录水平随年龄的显著升高。有趣的是,与年龄匹配的对照相比,在ad病例中npc1和naglu转录水平也显著较高。那些结果支持本文所述研究的可行性,并表明由alp基因中的功能性杂合变体导致的单倍体不足性会影响发展ad的风险,可能会影响app代谢、aβ生成以及体外和体内aβ降解。
[0438]
(i)鉴定ad中富含罕见功能变体的alp基因
[0439]
在4,000例ad病例和5,000名内部对照中,对所有alp基因(n=430)进行了单变体和基于基因的分析。在来自阿尔茨海默病测序项目(adsp)的独立样品(5,000例ad病例和4,500名对照)中复制了这些结果。使用来自外显子组聚合联盟(exac)数据库的非芬兰欧洲人(n=33,350)的wes数据来分析所有alp基因,以确定来自欧洲血统的个体的alp的每个基因中的基线遗传变异。
[0440]
(ii)体外确定alp的所选的候选基因对app代谢、aβ生成和aβ降解的功能效应
[0441]
生物化学和基于细胞的测定被用于验证所选的变体对蛋白功能、蛋白稳定性的影响及其对alp功能的影响。在原代神经元中检查了经验证的功能变体对app转运、app半衰期、app加工机制和aβ生成的影响。在来自缺陷或半合子小鼠(naglu、npc1和dnajc5基因)的神经胶质细胞中测试了aβ摄取和降解。
[0442]
(iii)确定所选的候选alp基因的单倍体不足性对老年小鼠中ad病理发展的功能作用
[0443]
可以确定在ad发病机制的早期(4个月)和晚期(8个月大),轻度的溶酶体损伤是否会加速5xfad小鼠中的aβ生成、斑块沉积、突触丢失和胶质增生。在没有fad突变的24个月龄小鼠中进行了遗传性慢性遗传性溶酶体损伤对ad相关表型的影响的定量病理学调查。
[0444]
这些研究形成了与ad相关联的alp功能障碍的遗传因素的原理论证基础。目前缺乏这方面的知识,并且可以产生新的治疗靶。
[0445]
研究策略
[0446]
意义
[0447]
自噬
‑
溶酶体途径(alp)是细胞内细胞器和易聚集蛋白的降解的主要途径。自噬(字面意思是“自我吞噬”)是一种细胞内降解途径,其负责经由溶酶体消化和回收营养物质。真正的功能性自噬反应指导内源性或外源性来源的细胞质材料在溶酶体中降解。溶酶体通过控制细胞清除和能量代谢的溶酶体至细胞核的信号传导机制在营养感测和信号传导途径中起重要作用,该信号传导途径涉及哺乳动物雷帕霉素复合物1(mtorc1)激酶复合物和转录因子eb(tfeb)的靶。alp是一种细胞内质量控制系统,该系统即使在没有任何疾病相关联突变蛋白的表达的情况下也对神经变性发挥保护作用。“核心”自噬基因(atg5和atg7)的神经元特异性缺失会导致异常蛋白的积累、进行性神经变性和过早死亡。
[0448]
ad中的alp功能障碍
[0449]
虽然家族形式的ad是由增加的淀粉样β(aβ)生成引起的,但对晚发型散发性ad患者的最近研究表明aβ的受损的清除。因此,产生与清除之间的平衡决定了aβ水平和形成淀粉样斑块的倾向。在人脑中的正常衰老期间,alp“核心”基因被转录下调。值得注意的是,与正常衰老相反,在ad患者的大脑中存在alp的转录上调,这可能代表了系统应对异常蛋白的积累的代偿性尝试。存在beclin 1(一种对alp中自噬体形成必不可少的多功能蛋白)水平的降低,rab5和rab7(调节囊泡沿内体
‑
溶酶体途径转运的小ras相关的gtpase(rab)蛋白)水平的升高,和散发性ad脑中巨噬细胞(高lc3
‑
ii水平)和mtor信号传导(磷酸化的p70 s6激酶)的异常激活以及营养不良神经突中自噬液泡(av)和溶酶体致密体的大量神经元积累。神经病理学研究还发现,ad脑中的自噬
‑
溶酶体病理有助于ad的发病机制,然而,潜在机制尚不十分明确。
[0450]
遗传性alp功能障碍加剧了ad病理
[0451]
在多种转基因小鼠ad模型中也已经发现alp的变化。beclin 1在两种ad小鼠模型(j20和t41;happ751v171i,km670/671nl)中的单倍体不足性对其溶酶体产生了进一步的破坏,促进了细胞内和细胞外aβ的积累,并加剧了神经变性。溶酶体神经氨酸酶1(neu1)的缺失加剧了ad模型中的aβ病理(5xfad;app km670/671,i716v,v717i/psen1m146l/l286v)。相比之下,neu1的过表达会降低ad病理。这些结果一起表明,alp“核心”基因或溶酶体蛋白的变化加重ad病理。
[0452]
改善alp功能会减少淀粉样ad相关表型
[0453]
app/ps1(appk670m/n671l/ps1m146l)小鼠在脆弱神经元群体中表现出异常的巨噬细胞活化,甚至在aβ的细胞外沉积之前。然而,在app/ps1小鼠模型中,神经元和星形胶质细胞中tfeb的靶向表达减少了aβ斑块。tfeb表达驱动多个溶酶体和转运基因的转录上调,并增加溶酶体酸化和功能。在ad小鼠模型(tgcrnd8;happk670n/m671l/v717f)中激活溶酶体半胱氨酸蛋白酶(通过删除半胱氨酸蛋白酶抑制剂(cystatin)b)挽救了自噬溶酶体病理,减少了异常aβ和泛素化蛋白的积累,减少了细胞外淀粉样蛋白沉积和总脑aβ40/42水平,并防止了学习和记忆测试缺陷的发展。在两种ad小鼠模型(j20和app/ps1)中,溶酶体蛋白酶的药理学激活降低了aβ42水平,改善了认知测试的表现和突触缺陷。在多种ad模型(j20;happ695,751,770v171f,km670/671nl),(3xtg
‑
ad;appswe/taup301l),(app
‑
ps1;appswe/psen1de9)中,alp的药理学激活(mtor抑制)改善了认知缺陷并减轻β淀粉样蛋白的积累。这些结果表明,在多种ad小鼠模型中,alp的整体激活或溶酶体蛋白水解的选择性增强均促进了淀粉样蛋白的清除。
[0454]
aβ在内体
‑
自噬
‑
溶酶体隔室中产生
[0455]
细胞研究表明,内体
‑
溶酶体系统是aβ产生的主要场所。然而,关于aβ产生的确切地点尚无共识。aβ是在体外和体内诱导大自噬后产生的。aβ的积累增加了mtor信号传导,而减少mtor信号传导降低了aβ水平,这表明alp活化与aβ水平之间的负反馈。在自噬激活下,自噬液泡成为具有最高γ分泌酶活性的细胞部位。psen2和呆蛋白(催化必需的γ分泌酶组分)位于溶酶体中。事实上,psen1调节溶酶体ph。体外溶酶体功能的药理学损伤导致aβ产生的变化。溶酶体ph的变化会减少aβ的分泌。溶酶体蛋白酶抑制剂减少淀粉样蛋白生成的app片段的产生。所有这些研究表明,整体溶酶体功能在正常和异常的app加工及随后的淀粉样蛋白生成中起重要作用。同时,在缺乏特异性溶酶体基因的小鼠中进行的研究揭示,每种基因在app加工和aβ产生中的独特作用。来自人类病理、小鼠和细胞模型的所有证据都强烈地表明,自噬诱导中的缺陷发生在疾病早期,但溶酶体清除缺陷发生在疾病的更晚期。然而,尚不清楚alp的变化是ad病理的原因、结果还是调节因素。
[0456]
人类溶酶体基因和ad的完全功能丧失
[0457]
目前对溶酶体基因与ad之间关系的理解是由关注溶酶体贮积病(lsd)中的ad病理的很少研究形成的。这些研究未在粘多糖贮积症(mps)、c型尼曼
‑
皮克病(npc)或神经元类蜡样脂褐质病(ncl)脑中发现aβ斑块。然而,mps、npc和ncl患者在整个大脑的细胞的胞质中表现出强烈的弥漫性aβ信号(参见,例如图13a和图13b)。与正常对照脑相比,mps患者表现出可溶性aβ水平的显著升高。aβ38、aβ40和aβ42的增加的csf水平表明在患有npc的患者的脑中增加的γ分泌酶依赖性aβ释放。与对照相比,患有ncl的人类患者表现出aβ40和aβ42水
平的显著降低(参见,例如图13a和图13b)。此外,在没有fad突变的过表达的情况下,npc1(尼曼
‑
皮克病)、cln3(巴顿病)和hexb(桑德霍夫病)基因缺陷的小鼠表现出细胞内app片段(α
‑
ctf/β
‑
ctf)和aβ40/42水平的升高。在idua(mps
‑
i)、sgsh(sanfilippo a)、gba(戈谢病)和tpp1(晚期婴儿巴顿病)基因缺陷的小鼠中,已经报告了细胞内app/aβ水平的升高,而无aβ斑块。在idua(mps
‑
i)、sgsh(a型sanfilippo病)基因缺陷的小鼠中发现,与未检测到aβ42的对照相比,aβ40水平的三倍增加。相反,在asah1(法伯病)或ppt1(婴儿巴顿病)基因缺陷的小鼠中发现细胞内app/aβ水平的显著降低。这些研究表明,即使没有aβ斑块,app的转运或加工在人患者和小鼠模型中都受到影响,具有溶酶体基因的完全功能丧失。有趣的是,人类和ad转基因小鼠的认知功能下降与aβ斑块负荷不成比例,但确实与可溶性aβ物种相关。来自转基因ad小鼠的数据表明,神经内aβ的神经毒性大于细胞外aβ。在人和小鼠ad模型中,细胞内aβ的积累已显示先于细胞外沉积。因此,除了异常生成aβ40或aβ42之外,人和缺乏溶酶体基因的小鼠的短寿命也可能阻止了研究人员发现aβ斑块。
[0458]
人alp基因的遗传变异
[0459]
在人类基因组中至少存在430个与alp相关联的基因(38个自噬基因,161个自噬调节基因,64个溶酶体基因和167个溶酶体调节基因)。所有alp基因中的38%(157个基因)的突变会导致人类孟德尔病(omim)。最近对60,000人中存在的突变的频率和类型的分析发现,大多数alp基因“不能耐受功能丧失”突变,并且携带的潜在有害变异比由中性进化模型预测的要少。这与敲除小鼠中大多数“核心”alp基因在胚胎发生或新生儿期的致死性及其对细胞维持和存活的重要性一致。研究最多且最熟知的alp基因是溶酶体基因,当突变时会导致lsd。有趣的是,这些引起lsd的基因中的大多数(≈50)对lof突变并不耐受,并且在人类中表现出相当大的遗传编码变异。因此,流行病学研究已经表明,在健康人中溶酶体酶活性的水平存在十倍范围的差异。与对照相比,携带导致多个溶酶体基因(包括gba、npc1、galc、gaa、gla和idua基因)中的单倍体不足性的遗传变体的个体表现出显著较低水平的酶活性。此外,npc1基因中的单倍体不足性会导致人类携带者中的其他显著的代谢异常。长期以来,人们一直认为携带alp基因的一个单一正常拷贝不会对健康造成影响。然而,大量的遗传证据支持gba基因中的功能变体作为发展帕金森病和路易体病的主要遗传风险因素的作用。在纯合子状态下导致儿童lsd(戈谢病)的相同变体(当以杂合方式存在时)会影响成人发作的神经退行性疾病的风险。这些研究表明,溶酶体基因中的单倍体不足性易导致成年人中常见的神经退行性障碍。缺乏对影响ad风险的alp基因中功能性杂合变体的贡献的系统性评估。
[0460]
创新
[0461]
本文所述的研究在概念上是创新性的,可系统且全面地评估与众所周知的与ad相关联的alp中的功能障碍相关联的遗传变异(参见,例如,第(i)节),以及了解与体外(参见,例如,第(ii)节)和体内(参见,例如,第(iii)节)ad相关联的alp基因中罕见功能性杂合变体的影响。此外,这些研究仔细地检查了神经元病理,以确定遗传性改变alp的后果及其对临床相关终点的淀粉样病理的影响。目前对ad中的alp基因中的遗传变异的理解主要是有限的研究,其报告了复制率非常低的分离群体中的虚假关联。本文所述的研究可以克服该限制,因为该设计在代表一般群体的非常大的样品中全面地评估了alp的每个基因中的遗传变异对发展ad的风险的贡献。因此,除了内部wes和外显子组芯片数据外,还使用了来自
两个大型公开可用的数据库的wes数据,即外显子组聚合联盟(exac)(n≈61,000)和阿尔茨海默病测序项目(adsp)(n≈10,000),用于总的ad病例(n≈4000)和对照(n≈5000)。使用这些多个数据集,设计了一组分析,其将解决与ad相关联的alp中众所周知的功能障碍的遗传结构。本文所述的研究并入了创新的综合框架,该框架将计算方法和实验数据相结合,以验证所选的alp基因在体外和体内的功能效应。基于细胞的测定由生化数据、活细胞测定、来自小鼠脑细胞类型特异性样品的rnaseq数据、人类ad病例和对照的全基因组基因表达数据、来自不同阶段人类ad病例的全基因组基因表达数据、来自不同年龄的人类的全基因组基因表达数据以及来自与aβ斑块相关的ad小鼠模型的全基因组基因表达数据补充。因此,作为这些数据挖掘工作的结果,不仅在原代神经元中,而且还在小胶质细胞中进行了第(ii)节中所述的实验。细胞类型特异性rnaseq数据显示,与在神经元中相比,在小胶质细胞中,大多数alp基因表现出较高的表达水平。本文所述的方法克服了之前关于alp基因在ad中的作用的非结论性研究。第(iii)节中描述的研究可以解决以下问题:年龄和易损脑区上的alp基因的单倍体不足性是否影响app加工和转运、aβ斑块负荷和aβ40/42水平。这些研究可以克服使用alp基因的敲除小鼠的先前研究的局限性,该alp基因的敲除小鼠表现出快速神经变性和短寿命,这使解释该缺陷基因对ad的影响变得复杂。这些研究通过合作创新是可能的,该合作创新涉及具有跨越神经遗传学、溶酶体生物学、lsd动物模型、细胞和小鼠模型中ad病理学的专业知识的研究人员。
[0462]
方法
[0463]
这里,使用最先进的基因组学工具和大型数据集揭示与ad相关联的alp功能下降背后的遗传结构。此外,所选的变体和基因对app代谢、aβ生成和aβ降解的影响可以在体外和体内被验证。
[0464]
数据和结果
[0465]
溶酶体基因中的杂合变体会影响发展ad的风险。
[0466]
通过挖掘公共数据库和文献中的现有注释获得的自噬
‑
溶酶体基因集(约430个基因)的列表已经在之前手动编制。可以分析ad组群中的所有alp基因。然而,其完全功能丧失(lof)突变导致lsd的溶酶体基因是在人和小鼠模型中研究得最好的导致神经变性的alp功能障碍模型。此外,对分离(地理和遗传)群体中单个溶酶体基因的小样品量研究支持了溶酶体基因中的遗传变体在ad风险中的作用。然而,这些研究尚未被复制。因此,通过使用大的数据集(该数据集很好地代表了具有欧洲血统的群体)聚焦于四十六个溶酶体基因来开始分析alp基因对ad的遗传贡献。目前可用的一系列生物信息学工具的缺点是,不存在预测未知功能的编码变体是否具有功能(生物学)后果的完美算法。因此,基于未知功能的编码变体与已知完全或接近完全lof变体(当它们为纯合的或复合杂合的时会导致lsd)之间的共同特征,定义了潜在功能变体的纳入和排除标准。
[0467]
发现样品由来自523例不相关ad病例和386名对照的wes数据组成。表5显示了与ad相关联的顶级溶酶体基因。如所预期的,来自exac数据集(欧洲人,非芬兰裔)的基因特异性积累等位基因频率(cmaf)与来自内部ad数据库(欧洲裔的ad数据库)的cmaf高度一致(r2=0.97)。在ad组群中,将符合纳入标准的每个溶酶体基因中的编码变体按基因进行核对:82%为错义变体,15%影响选择性剪接,并且3%为无义突变。将罕见蛋白改变变体(cmaf)的负荷与在对照和exac中观察到的负荷进行比较。对于大多数基因,与对照相比,在病例中
存在过多的变异,但仅发现与sgsh基因(p=4.2
×
10
‑3;or=3.7,95%ci 1.4
‑
9.6)和cln8基因(p=1.0
×
10
‑2;or=8.9,95%ci 1.1
‑
68.1)的名义关联(参见,例如表5)。
[0468]
当与exac样品的cmaf相比时,十四个基因通过了非常严格的多重检验校正阈值p<1.0
×
10
‑4(0.05/450),并且十三个引起lsd的基因通过了基因水平显著性阈值p<2.4
×
10
‑6(0.05/20,000)(参见,例如表5)。接下来,使用adsp组群来复制表5中所列的发现,包括5045例ad病例和4500名对照(参见,例如表7)。
[0469]
表7.在adsp中引起lsd的基因中发现的罕见变体(复制样品)
[0470]
基因cmaf adcmaf nfeor95%cip值galns0.00040.00021.71.5
‑
2.02.35e
‑
14ctns0.00020.00011.91.6
‑
2.31.04e
‑
11mfsd80.00010.00012.71.9
‑
3.71.97e
‑
10gnptab0.00030.00021.51.3
‑
1.82.12e
‑
08tpp10.00020.00011.61.3
‑
1.91.48e
‑
06naglu0.00020.00011.51.1
‑
1.93.40e
‑
05smpd10.00040.00031.31.1
‑
1.42.79e
‑
04npc10.00020.00021.21.1
‑
1.43.000e
‑
04cln80.00020.00011.91.3
‑
2.63.35e
‑
04dnajc50.00050.00031.61.2
‑
2.83.40e
‑
04hexb0.00050.00041.31.1
‑
1.56.61e
‑
04
[0471]
在该独立样品中,九个基因复制了与ad的关联(参见,例如表7)。值得注意的是以下事实:在复制样品中发现的关联在相同方向上并且效应大小相同。
[0472]
随年龄和ad状态的npc1转录水平
[0473]
根据上述标准,选择了在两个样品中复制的三个溶酶体基因用于功能分析:编码细胞内胆固醇转运体1(npc1)的基因、编码n
‑
乙酰基
‑
α
‑
氨基葡萄糖苷酶(naglu)的基因和编码半胱氨酸串蛋白α(cspα)的dnajc5基因。npc1将低密度脂蛋白转运至晚期内体/溶酶体隔室,在该隔室中它们被水解并作为游离胆固醇释放。该基因中的lof导致c型尼曼
‑
皮克疾病。与在神经元中相比,npc1转录物在小胶质细胞和星形胶质细胞中表现出较高的表达水平(约4.5倍)。在神经病理学正常的人脑样品中,随着年龄的增长,npc1转录水平高度显著增加(p<0.0001)(参见,例如图10a)。与年龄匹配的对照相比,在ad病例中npc1转录水平显著更高(p=0.01,参见,例如图10b)。
[0474]
随年龄、ad状态和ad小鼠模型中的naglu转录水平
[0475]
naglu降解硫酸乙酰肝素,并且该基因的总lof导致iiib型粘多糖贮积症(mps
‑
iiib),也被称为sanfilippo综合征b。来自小鼠的脑细胞类型的rnaseq数据显示,与在神经元中相比,naglu转录物在小胶质细胞中表现出较高的表达水平(约20倍)。在神经病理学正常的人脑样品中,naglu转录水平随着年龄的增加而显著增加(p=0.02)(参见,例如图1a)。与年龄匹配的对照相比,在ad病例中naglu转录水平显著升高(p=0.007)(参见,例如图1b)。与野生型小鼠中的水平(黑线,参见,例如图1c)相比,在ad小鼠模型的皮质中(app,p.k670n/p.m671l/psen1,p.m146v;半合子的[het]或纯合子的[ho];参见,例如图1c),随着ad病理的发展,naglu转录水平也表现出成比例的年龄依赖性升高(右图,参见,例如图1c)。
[0476]
随年龄、ad状态和ad小鼠模型的dnajc5转录水平
[0477]
cspα是一种突触伴侣蛋白,其参与突触的胞吐作用和蛋白稳态的维持。cspα中的杂合突变导致常染色体显性成人发作神经元蜡样脂褐质沉积症(ancl)。dnajc5转录产物在神经元和最易受ad病理影响的脑区域中被高度表达。在来自额叶皮质区的神经病理学正常脑样品中发现dnajc5转录水平随年龄的降低(参见,例如图8a)。在ad和对照的激光捕获微解剖的非缠结神经元中,与年龄匹配的对照相比,在ad病例中dnajc5转录水平显著较低(p=<0.0001,参见,例如图8b,左图)(geo数据库;gse5281系列)。在不同的研究中重复了这一发现(geo数据库;gse15222系列)(参见,例如图8b,右图)。还发现,与野生型小鼠中的水平(参见,例如图8)相比,在ad小鼠模型(app,p.k670n/p.m671l;图8)的皮质中,dnajc5转录水平表现出年龄依赖性降低,并且与ad病理的发展成反比(参见,例如图8)。所有这些结果表明,npc1、naglu和cspα参与了ad的发病机制。
[0478]
内源性cspα定位于溶酶体中,并且突变的cspα影响自噬蛋白水平(lc3
‑
ii p62)
[0479]
内源性cspα与溶酶体标记物共同定位于原发性皮质神经元和神经元样细胞类型(n2a)的胞体、神经突和突触结中(参见,例如图12a)。亚细胞分级显示,相当比例的cspα与另一种溶酶体标记物(lamp1)共沉积(参见,例如图12b)。这些结果表明,内源性cspα是一种溶酶体相关联蛋白。引起ancl的突变(p.l115r)的表达导致高分子量cspα聚集,以及溶酶体蛋白lamp1和snap23水平的升高。存在p62的减少和从lc3
‑
i向lc3
‑
ii的持续转化,这表明自噬的激活以及自噬体和溶酶体的融合受阻(参见,例如图12c)。与空载体相比,这种引起ancl的突变显著地增加了溶酶体跟踪器信号的水平。相比之下,cspα
‑
wt的过表达显著地降低了溶酶体跟踪器信号(参见,例如图12d)。
[0480]
cspα影响体内app加工
[0481]
ancl患者的大脑未出现aβ斑块或神经原纤维缠结。然而,在ancl患者的皮质神经元中存在显著的app/aβ细胞内积累(ancl,参见,例如图13a)。对来自ancl、ad和健康对照样品的脑样品的洗涤剂可溶和不可溶(胍)级分中的aβ进行的定量揭示,与对照和ad样品相比,ancl患者中的aβ40和aβ42水平显著降低(参见,例如图13b)。这些结果表明,cspα在aβ生成和ad发病机制中起作用。
[0482]
研究设计与方法
[0483]
(i)鉴定ad中富含罕见功能变体的alp基因
[0484]
由alp的基因中功能性杂合变体引起的单倍体不足性影响发展ad的风险的假设可以被测试。如本文所述,将大数据集(内部数据库和公共可用的数据集)中对alp基因的预测罕见功能变体的分析相结合的综合方法用于优先考虑alp中有证据表明参与ad风险的候选基因。
[0485]
第(i)节的方法学和分析:定义罕见变异的等位基因频率阈值
[0486]
初步分析集中于溶酶体基因。相同的方法适用于所有alp基因。然而,在其余alp基因中没有由lof突变引起的人类疾病的情况下,根据溶酶体基因的经验调整了纳入标准。存在在ncbi clinvar数据库中报告的约2740种引起lsd的变体。大多数ad样品均为欧洲血统。因此,为了在具有相似遗传背景的群体中确定溶酶体基因中的遗传变异的基线,从exac中选择了非芬兰样品(约33,000个个体)。在exac样品中注释为杂合的测试的溶酶体基因中发现了288种lof变体:76%为错义变体,10%影响替代剪接,并且12%为无义突变。通过sift
预测,大多数lof变体是有害的(87%),并且通过polyphen2预测,大多数lof变体是危险的(84%)。大多数(73%)lof变体位于高度保守的核苷酸中(grep评分>4)。在ncbi clinvar数据库中,exac中报告的几种lof变体未被归类为引起lsd的变体。在每个测试的溶酶体基因中均发现了杂合lof变体,但杂合lof变体的数量与hyal1基因中发现的数量不同,与arsa基因中发现的二十种不同。每个基因的这些杂合lof变体的cmaf范围从cstd基因中的1.5
‑
05到npc2基因中的0.003。因此,采用1
×
10
‑3的cmaf阈值作为保守上限。使用从lof变体的分析中获得的信息(包括lof变体的最高maf、sift、polyphen2或gerp评分),定义了以下纳入标准:1)ad病例中的调用率>98%,2)每个变体的maf<0.01%,3)由exac或系综注释为错义的指定规范转录物中的仅可能的蛋白改变变体,4)框架移位,5)无义,6)影响剪接供体和受体区域的变体,和7)如果在ncbi clinvar数据库中有致病性证据并且位于3'或5'utr区域。在exac中未发现的变体,或ncbi clinvar数据库中不存在的同义、内含子、3'或5'utr变体或maf>0.01%、exac和clinvar中的错误调用被排除在外。为了确保群体特异性变体对本分析没有混杂效应,根据主组分结果选择了个体。
[0487]
表8示出了可用于进行遗传分析的样品的汇总。
[0488]
表8.阿尔茨海默病数据集。*目前正在对adsp复制数据集进行测序
[0489][0490][0491]
可以访问来自knight阿尔茨海默病研究中心(knight adrc)、阿尔茨海默病神经影像学倡议(adni)、nia
‑
load研究、西班牙数据集和阿尔茨海默病测序项目(adsp)的超过17,000个个体的表型数据、dna和/或遗传数据。此外,目前正在对来自adsp的10,000个样品进行测序(wgs)。可以分析内部数据库中的所有alp基因。这些数据集的描述先前已发布。使用与国家神经病学和交流障碍和中风
‑
阿尔茨海默病和相关障碍协会对可能的ad进行的标准等效的标准,每个病例都接受了阿尔茨海默病类型痴呆的诊断。对照接受与病例相同的评估,但认知正常。所有个体都是欧洲血统,并获得了来自所有参与者的书面同意。
[0492]
阿尔茨海默病测序项目(adsp)
[0493]
来自ad病例和对照的wes数据用于对alp基因执行本文所述的所有遗传分析。数据于2015年12月从adsp下载。发现阶段数据集含有来自113个家庭的584名受试者的wgs数据和超过4000名受试者的系谱数据;5096例病例和4965名对照的wes数据,以及来自受ad多重影响的家庭的另外853例受试者(682例[510例非西班牙裔,172例西班牙裔])和171名西班牙裔对照受试者的全外显子组序列数据。adsp复制阶段已经在进行中,并且包括另外的10,000个个体的wgs数据。
[0494]
公开可用的wes数据
[0495]
exac中包括的非芬兰欧洲人样品的等位基因频率被用作所有alp基因分析的等位
基因频率参考。数据下载自exac(版本0.3.1,2015年3月)。在分析中仅包括来自具有覆盖至>30
×
的中位序列深度的高比例的编码区以及仅高质量(带通滤光器)变体的alp基因的数据。
[0496]
全外显子组测序数据收集的技术说明
[0497]
存在来自2,000个个体的wes。使用sureselect 52mb靶富集系统(安捷伦)进行外显子组富集。dna通过配对末端读数(illumina hiseq2000)来测序。使用novoalign和sam工具执行比对和变体调用。一旦数据被处理,并且通过外显子组测序检测到的序列变体通过基因分型被确认,就应用已被开发并用于其他外显子组测序项目的高效和有效的用于数据管理、质量控制、清洁、注释和分析的方法。
[0498]
统计检验
[0499]
为了适应具有适中的效应大小的罕见变体的效应,使用了已开发的经验证的统计方法来分析与罕见变体的关联。简而言之,基于基因的方法将区域内的罕见变体折叠成单个值,并且然后测试区域内的罕见变体与感兴趣性状之间的关联。序列核关联检验(skat)用于检验基因区域内的状态与罕见变体之间的关联。skat可以解释同一基因中在不同方向上具有影响的变体,并对混杂的协变量(包括群体标记物)进行调整。然后,独立的病例对照样品被用于复制来自发现数据集的发现。对每个不同数据集的分析是分别进行的。进行联合分析以合并p值和or。该方法已被成功地用于先前的研究中,以鉴定ad的新基因。
[0500]
群体结构
[0501]
该分析定义了分析中包括的内部样品的欧洲血统。eigenstrat与hapmap样品一起在样品上用作锚,以确认自我报告的种族/民族。
[0502]
生物信息学分析
[0503]
以下公开可用的数据库被用于补充分析:在线人类孟德尔遗传(omim)、exac、gwas目录、clinvar数据库和人类自噬数据库。
[0504]
功效分析
[0505]
为了确定检测与ad相关联的遗传变体的功效,在sas中使用proc power运行分析。使用在0.01至0.50范围内的次要等位基因频率、1.2至3.6的or以及4,000至7,000个样品量运行分析。对于单变体分析,α被调整为5
×
10
‑8;并且对于基于基因的分析,α被调整为5
×
10
‑6。存在大约80%的功效以检测or>1.19(或<0.84)的效果。
[0506]
预期的结果
[0507]
预计通过本文所述的研究来揭示ad风险与alp基因的新的关联。研究已经表明,可以鉴定alp中与ad风险相关联的新基因(参见,例如表5),并且这些基因可以在具有适当覆盖深度的独立样品中被复制(参见,例如表7)。与在大的数据集(exac)中大多数“核心”alp基因中发现的最小遗传变异一致,除了不存在与其相关的人类疾病外,预计在ad患者中的“核心”alp基因中没有功能变体的富集。
[0508]
替代方法
[0509]
结果和功效计算表明,在基于基因的分析中,存在足够的功效以检测>2.7的平均优势比。如果alp基因的关联未能与病例对照设计复制,则可以使用内表型设计。因此,可以通过对alp基因和以下每种csf生物标记物(t
‑
tau、p
‑
tau和aβ42)进行单变体和基于基因的分析,来确定alp基因中的变体对ad的csf生物标记物水平的影响。为了评估alp基因的调控
基因组区域是否可能参与发展ad的风险,可以分析在来自由国际阿尔茨海默病基因组学项目(i
‑
gap)先前发表的gwas的数据中alp基因的关联,该数据由总共25,580例ad病例和48,466名对照组成。一种补充方法可以是检查alp基因是否影响发作年龄(aao)。
[0510]
剩余的约384个alp基因的遗传变异及其与ad风险的潜在关联的分析已经完成。可以使用adsp复制阶段增加样品量,其包括另外10,000个个体(ad病例和对照)的wgs数据。与基因组聚集数据库(gnomad)的组织者建立了适当的合作关系。gnomad是exac的扩展,其含有来自123,136个个体的外显子组序列数据和来自15,496个个体的全基因组测序,以在更大的数据集中复制这些发现。adsp数据被包括在gnomad中,这排除了使用gnomad的公共版本用于当前的分析。
[0511]
(ii)(a)体外确定alp的所选的候选基因对app代谢、aβ生成和aβ降解的功能作用
[0512]
评估在第(i)节中鉴定的与ad相关联的每种alp基因的所有变体超出了本文所述研究的范围。因此,优选考虑在第(i)节中鉴定的naglu、npc1和dnajc5基因的前3
‑
5个变体。根据ad患者中的频率、通过sift和polyphen2预测的对蛋白的影响以及gerp保守评分来定义顶级变体。根据来自发现和复制样品的数据强度来选择这些基因。推测与ad风险相关联的新型alp基因会影响app代谢、aβ生成和体外aβ降解。
[0513]
评估对蛋白产品的影响
[0514]
已经选择了共享lof变体的许多计算机特性的变体。本文概述的实验可以生成实验数据以验证对其相应的蛋白的功能作用。使用naglu、npc1和dnajc5基因的cdna的定点诱变对选定的变体进行工程改造。如前所述,将野生型和突变型cdna亚克隆到慢病毒载体中。慢病毒载体按照涉及重组或合成核酸分子的nih研究指南的第iii
‑
e
‑
1节在bsl2设施中生产、处理和处置。如前所述进行原代神经元培养。naglu、npc1和dnajc5基因在来自缺陷小鼠的原代神经元中表达,并且通过蛋白免疫印迹来定量对蛋白稳定性的影响。通过免疫荧光共聚焦图像来评估突变的蛋白的亚细胞定位,以在溶酶体(lamp
‑
1或
‑
lamp
‑
2)、早期内体(eea1)、晚期内体(rab7)、er(kdel)和高尔基体(giantin)标记物中共同定位所选的蛋白。如前所述,使用荧光测定法来测试naglu变体对酶活性的影响。所选的变体对npc1的影响如先前公布的进行。通过所选的变体防止snap
‑
25降解的能力来测试所选的变体对dnajc5的功能作用。
[0515]
评估对app转运、内吞作用和亚细胞定位的影响
[0516]
多项研究已经表明,app的内吞作用对于其在app淀粉样变性途径中与内体和多囊体内的β分泌酶和γ分泌酶共同定位是至关重要的。继发于溶酶体功能障碍的内体通量的损伤导致在该细胞器内的增加的转运时间,这增加了β裂解和γ裂解的倾向,并且因此增加了aβ的生成。为了确定naglu、npc1和dnajc5基因中的选定变体是否影响app的稳态水平、app内吞作用或app进入溶酶体用于降解的增强的通量,可以使用如先前公布的细胞表面生物素化测定法来确定细胞内app出现的动力学和细胞表面中app的水平。在用蛋白合成抑制剂环己酰亚胺处理0、5、10、30分钟时,通过蛋白免疫印迹来测量对全长app半衰期的影响。进行免疫组织化学共定位显微镜技术以研究对app和sorl1亚细胞定位的影响。分别通过蛋白免疫印迹和rt
‑
qpcr来测量app加工机制的蛋白和转录水平,包括α分泌酶(adam10和adam17)、β分泌酶1(bace1)和γ分泌酶复合物(psen1和呆蛋白)。
[0517]
评估对aβ生成的影响
[0518]
很大一部分app被靶向溶酶体,并且在存在溶酶体酸化抑制剂的情况下,app水平在细胞中迅速升高,这表明溶酶体降解会驱动app蛋白水解,以阻止aβ肽的形成。此外,与正常对照脑相比,人类sanfilippo患者(naglu缺陷)呈现出可溶性aβ水平的显著升高。据报告,在具有npc(npc1缺陷)的患者的脑中,aβ40和aβ42的csf水平增加,并且β裂解的可溶性app水平没有变化。与对照相比,具有dnajc5突变的人类患者表现出aβ40和aβ42水平的显著降低。因此,可以评估所选的变体是否影响细胞培养物中aβ的生成。通过如先前公布的夹心elisa来检测细胞裂解物和细胞培养基中的aβ物种。简而言之,用小鼠单克隆包被抗体hj2(抗aβ35
‑
40)和hj7.4(抗aβ37
‑
42)捕获aβx
‑
40肽和aβx
‑
42肽。hj5.1(抗aβ13
‑
28)(一种靶向中心结构域的生物素化抗体)或hj3.5(抗aβ1
‑
13)(其靶向n
‑
末端氨基酸)被用作检测抗体,然后是链霉亲和素
‑
聚
‑
hrp
‑
40(菲茨杰拉德工业公司)。通过蛋白免疫印迹来测量app衍生的蛋白水解片段,诸如α
‑
ctf和β
‑
ctf以及sappα和sappβ。还通过蛋白免疫印迹来监测全长app的水平。
[0519]
评估对aβ降解的影响
[0520]
小胶质细胞在淀粉样斑块周围增殖,并且吞噬淀粉样材料,但随后的降解受损,导致ad中淀粉样物质的逐渐积累。小胶质细胞为何可以摄取原纤维aβ但不能降解原纤维aβ尚不清楚。然而,来自ad患者的小胶质细胞显示beclin
‑
1的减少和随后的alp功能障碍。此外,不溶性原纤维aβ影响氯离子通道cic
‑
7向原发性小胶质细胞中的溶酶体的转运,这损害溶酶体降解。然而,恢复溶酶体酸化会增强aβ的降解。总之,该证据表明小胶质细胞中的alp不足性可能有助于ad发病机制。这里介绍的数据挖掘工作揭示了,与在神经元中相比,naglu和npc1基因在小胶质细胞中呈现出较高的表达水平。因此,可以评估来自用选定的变体转导的naglu和npc1缺陷和半合子小鼠的原代小胶质细胞是否可以摄取和降解外源性aβ。aβ的摄取和aβ的降解如先前公布的进行。
[0521]
评估对alp功能的影响
[0522]
可以测试来自半合子小鼠的神经元是否表现出alp功能障碍,以及这些变化是否通过所选的变体而增加。lc3和p62的蛋白免疫印迹被用作巨噬细胞活化的间接指标。在不存在或存在自噬系统的激活剂(雷帕霉素和torin1)、自噬抑制剂(巴弗洛霉素a1)、溶酶体营养剂(氯喹、氯化铵)和e64/亮抑蛋白酶肽的情况下,通过细胞中存在的lc3
‑
ii的量来评估自噬通量。通过使用mcherry
‑
gfp
‑
lc3标记物的活细胞成像对此进行补充。可以通过lc3和lamp1的共定位来进一步评估自噬体
‑
溶酶体融合。溶酶体跟踪器用于定量每个细胞的酸性隔室的数目。
[0523]
生物信息学分析
[0524]
以下公开可用的数据库用于补充分析:基因表达综合数据库;脑rna
‑
seq2;来自小鼠痴呆网络(小鼠demnet)的基因表达数据;polyphen2;sift;小鼠基因组信息学。
[0525]
预期的结果
[0526]
预计在第(i)节中鉴定的alp基因风险变体会导致部分功能丧失并改变alp功能。预计可检测出这些蛋白的功能中5
‑
20%的残留活性。还预计选定基因的过表达或下调会影响app转运、app代谢、aβ生成或aβ降解。此外,预期本文所述的实验还允许评估alp基因中的选定变体对神经元和小胶质细胞存活的影响。
[0527]
如果在naglu、npc1和dnajc5基因中未观察到wt与风险变体之间的显著差异,这可
能是由于过表达掩盖了功能的微妙变化。引起在一生中可能在疾病中表现出来的微小变化的ad风险变体对于在细胞培养的日子里看到影响可能是一个挑战。可替代地,可以使用来自5xfad转基因小鼠或n2a695细胞的原代神经元,并用naglu、npc1和dnajc5基因中的选定变体进行转导,并测试对aβ生成的影响。
[0528]
在体外鉴定naglu、npc1和dnajc5基因中影响ad的风险和发病机制的变体后,接下来的步骤是利用直接从来自人类的成纤维细胞产生ipsc和基因组编辑方法的进展。可以使用来自携带naglu、npc1和dnajc5基因中的变体的ad患者的ipsc衍生神经元或神经胶质细胞,以将此类变体对app代谢的影响与等基因crispr校正的细胞进行比较。一旦这些工具到位,就可以利用诸如酶替代(naglu)、环糊精(npc1)或alp的药理学调节的治疗策略,并在体外测试它们对aβ生成或aβ降解的影响。
[0529]
(ii)(b)确定所选的候选alp基因的单倍体不足性对老年小鼠中ad病理的发展的功能作用
[0530]
虽然已经表征了小鼠和人类中naglu和npc1基因的完全功能丧失的神经退行性后果,但对这些基因的单拷贝的长期后果知之甚少。一直以来,人们都认为半合子小鼠和人类是正常的。然而,最近表明,npc1基因中的单倍体不足性会导致人类和小鼠中的显著的代谢异常。假设ad病理由较温和形式的遗传性alp功能障碍发展而来,并且其出现可能需要额外的年龄相关alp损伤。主要终点为在小鼠中在4个月龄时(在斑块沉积前)测得的可溶性aβ水平和在8个月龄时在存在引起fad的突变中的斑块负荷。对可溶性aβ水平的影响是24个月大的naglu、npc1和dnajc5半合子小鼠中的主要终点。半合子小鼠由市售缺陷小鼠产生。
[0531]
对ad病理的小鼠模型的影响
[0532]
在具有sanfilippo疾病的人类患者中,naglu蛋白功能的完全丧失导致可溶性aβ的水平比正常对照脑显著升高三倍。据报告,在完全丧失npc1功能的患者中,aβ40和aβ42的csf水平增加,并且β裂解的可溶性app水平没有变化。在ad小鼠模型的皮质中,随着ad病理的发展,naglu转录水平表现出成比例的年龄依赖性增加(参见,例如图1c)。在ad小鼠模型的皮质中,dnajc5转录水平表现出年龄依赖性降低,并且与ad病理的发展成反比(参见,例如图8a)。与对照相比,在dnajc5中具有杂合突变的人类患者表现出aβ40和aβ42水平的显著降低(参见,例如图13)。与ad转基因小鼠一样,人类的认知能力下降与aβ斑块负荷不成比例,但确实与可溶性aβ物种相关。鉴于来自人类lsd患者和lof小鼠模型的数据支持这些基因在细胞内aβ生成中的作用,可以确定在良好表征的ad小鼠模型中,轻度溶酶体损伤(naglu、npc1和dnajc5中的半合子)是否会加速aβ生成,该ad小鼠模型携带有利于aβ生成的fad突变。5xfad模型是一种侵袭性很强的淀粉样蛋白沉积模型,其在1.5个月时出现神经内aβ42,在2个月时出现斑块,在4个月时出现突触标记物的丢失和记忆缺陷,并且在9个月时出现神经元丢失。斑块的发展伴有反应性胶质增生。为了进一步确定naglu、npc1和dnajc5单倍体不足性是否会加剧现有的淀粉样变性过程,将naglu、npc1和dnajc5小鼠与5xfad转基因小鼠杂交。为该实验产生4组(30只小鼠/组)。在5xfad/naglu(+/
‑
)、5xfad/npc
‑
1(+/
‑
)和5xfad/dnajc5(+/
‑
)小鼠中测定了基因剂量对4个月和8个月时app代谢、aβ斑块负荷和aβ40/aβ42水平的影响。在5xfad小鼠中,四个月是淀粉样斑块沉积的早期时间点。8个月代表当淀粉样斑块丰富时的年龄。
[0533]
样品量
[0534]
样品量计算表明,在研究同等数量的雄性和雌性小鼠时,需要至少n=15只小鼠/组来检测诸如斑块负荷(sd=30%,α=5%)以及洗涤剂可溶性和不可溶性的aβ40和aβ42的终点的20%增加(具有80%功效)。将来自每个基因的与5xfad[5xfad/naglu(
‑
/+);15 5xfad/npc1(
‑
/+)和5xfad/dnajc5(
‑
/+)]小鼠杂交的十五(15)只半合子小鼠以及另外15只5xfad小鼠在4个月和8个月大时麻醉并处死,以采集大脑用于组织学和生物化学研究。
[0535]
溶酶体基因的单倍体不足性对老年小鼠的影响
[0536]
ad病理(例如,aβ斑块)通常是年龄依赖性的。然而,已发表的研究尚未解决年龄与alp功能障碍之间的相互作用。大多数在体内评估alp在ad中的作用的研究使用了药理学方法,或者完全没有alp基因和短期终点。采用遗传学方法以用于降低naglu、npc1和dnajc5基因的内源性水平,并对遗传性慢性遗传性溶酶体损伤对涉及aβ的ad相关表型的影响进行定量病理学调查。结果已经显示,在正常人脑样品中,随着年龄的增长,npc1和naglu转录水平有非常显著的增加(参见,例如图10a和图1a)。此外,与年龄匹配的对照相比,在ad病例中npc1和naglu转录水平显著较高(参见,例如图10b和图1b)。这些结果表明,对来自溶酶体基因npc1和naglu的聚集蛋白的代偿性反应是正常衰老过程的一部分。在ad模型中发现的异常升高表明,它们试图控制aβ的异常水平。相比之下,在神经病理学正常的脑样品中,dnajc5转录水平随年龄而降低(参见,例如图8a),并且与年龄匹配的对照(参见,例如图8b)相比,在ad病例中dnajc5转录水平显著较低。dnajc5编码一种神经保护性突触伴侣,其突变会损害alp功能(参见,例如图13b)。因此,那些基因的单倍体不足性可能会加重老年小鼠中的ad相关表型。
[0537]
样品量
[0538]
来自每个基因的十五(15)只半合子小鼠[naglu(
‑
/+);npc1(
‑
/+)和dnajc5(
‑
/+)]和15只野生型小鼠在24个月大时被麻醉并处死,以采集脑用于组织学和生物化学研究。
[0539]
淀粉样斑块的定量和aβ生成
[0540]
固定的冷冻脑切片(50μm)在小鼠的亚组群中用x
‑
34进行染色,并用hj3.4(抗aβ)抗体进行免疫染色,以量化斑块负荷(以%面积表示)。将来自对侧半球的脑组织匀浆中的aβ水平分级为可溶性(pbs)和不溶性(5m胍)级分,并使用elisa进行定量。评估了所选的基因的单倍体不足性对app加工机制的影响。
[0541]
突触标记物
[0542]
突触丢失是人类和ad小鼠模型中的常见发现。可以通过蛋白免疫印迹(使用针对突触前标记物:snap
‑
25、囊泡相关联膜蛋白2、突触结合蛋白1和突触素的抗体,如前所发布的)来评估所选的基因的单个拷贝是否会加速5xfad小鼠中的突触丢失。
[0543]
alp功能障碍
[0544]
使用如上所述的脑切片,用抗lamp1、lc3和p62抗体对切片进行免疫染色。
[0545]
对神经营养不良和反应性胶质增生的影响
[0546]
使用网状蛋白
‑
3(rtn
‑
3)抗体(rtn
‑
3选择性地积累在营养不良的神经突中)对来自上述组群的固定的冷冻脑切片进行免疫染色,以定量营养不良的神经突,如先前所公布的。先前的研究已经表明,naglu、npc1和dnajc5缺陷小鼠表现出增加的星形胶质增生。因此,在平行研究中,脑切片用抗cd11b和抗gfap抗体染色,以检查所选基因的一个单个拷贝对反应性胶质增生的影响。
[0547]
预期的结果
[0548]
预期naglu、npc1和dnajc5基因的半合子小鼠会加速和恶化5xfad小鼠中的aβ斑块负荷的负荷。预计所选基因的单倍体不足性会影响老年小鼠中的app代谢和aβ生成,从而增加突触丢失和反应性胶质增生,而不会生成aβ斑块。
[0549]
如果在半合子小鼠的脑中未发现app代谢和aβ生成的变化,则生成携带在第(ii)节所述的实验中验证的最显著变体的aav2/9载体,并立体定向地注射到新生半合子小鼠的海马中,并评估ad病理的存在。可替代地,通过注射携带针对naglu、npc1和dnajc5基因的经验证的shrna/rnai的aav2/9载体,在新生的5xfad转基因小鼠中敲除这些基因的转录物。crispr技术可以用于在选定的基因中产生对体外测定具有最大影响的变体的敲入小鼠。
[0550]
结合来自本文所述研究的结果以及naglu活性的荧光测定法和npc1生物标记物的质谱测定法的可用性,可以筛选大组群的ad病例和对照的csf、血浆、血清或干血点,以检测可以用作ad的生物标记物的特定缺陷。
[0551]
实例4:确定naglu中的遗传变异对阿尔茨海默病(ad)和帕金森病(pd)发病机制的影响
[0552]
本实例描述了naglu基因中的遗传变异在阿尔茨海默病(ad)和帕金森病(pd)发病机制中的体外和体内作用。
[0553]
越来越多的证据表明阿尔茨海默病(ad)、伴有路易体的痴呆和伴有帕金森病(pd)的额颞叶痴呆(ftd)之间的临床、病理和遗传重叠。在各种生化和细胞研究中,异常的硫酸乙酰肝素(hs)代谢正在成为ad和pd的常见致病机制,然而,潜在的机制尚未清楚。尚未对参与hs的溶酶体降解的酶的遗传变体在ad或pd的发病机制中所起的作用进行系统评估。这里,在大病例
‑
对照ad和pd组群中进行了遗传分析,并且在负责硫酸乙酰肝素(hs)降解的溶酶体酶基因中发现了罕见功能变体的显著富集,具有显著的效应大小。硫酸乙酰肝素蛋白聚糖(hspg)调控多种致病蛋白(包括淀粉样蛋白(aβ)和α
‑
突触核蛋白(α
‑
syn))的寡聚化、清除、内吞作用和转运。对致病蛋白的hspg结合的药理学抑制和对hspg合成的遗传性减少促进了致病蛋白的清除并减少了它们的聚集。到目前为止,尚不清楚n
‑
乙酰基
‑
α
‑
氨基葡萄糖苷酶(naglu)活性的降低以及所产生的hspg积累是否会影响app代谢、aβ斑块负荷或α
‑
syn聚集和扩散。本文所述的研究可以确定naglu基因中的亚形错义变体对aβ前体蛋白(app)转运、神经元中的aβ生成以及通过神经胶质细胞的aβ降解的影响。描述了一种在小鼠中模拟与人类神经退行性疾病相关的遗传变体的创新方法。使用嗜神经腺相关病毒(aav)载体,该载体能够在naglu半合子小鼠中在生命早期注射无创、广泛分布和持久的全神经表达亚形naglu变体,并进行长期随访。因此,不仅可以研究naglu变体的细胞效应,而且还可以研究衰老和杂合性的影响,如在ad和pd患者中所发现的。还可以确定naglu的减少或过表达是否影响良好表征的ad小鼠模型中存在的ad病理。可以确定naglu基因中的亚形错义变体是否影响原代神经元中α
‑
syn的结合、内化和聚集,以及那些变化是否通过底物减少、重组酶替代或基因疗法得到挽救。最后,可以使用一种可靠且良好建立的病理性α
‑
syn扩散的方法来确定亚形naglu变体是否会影响磷酸化的α
‑
syn的聚集体的形成、连通性依赖性扩散及其对疾病进展和寿命的影响。本文所述研究的结果对鉴定具有遗传决定的溶酶体功能障碍的pd和ad患者具有重要意义,其中此类功能障碍的恢复可以提供有效的疗法。
[0554]
本文所述研究的目的是在体外和体内确定naglu基因中的遗传变异在阿尔茨海默
病(ad)和帕金森病(pd)发病机制中的作用。这些研究并入了在小鼠中模拟与人类神经退行性疾病相关联的遗传变体的创新方法。本文所述的研究可以揭示与ad和pd相关联的新溶酶体基因,并且对ad和pd发病机制中溶酶体功能障碍的机制提供更深入的了解。
[0555]
越来越多的证据表明阿尔茨海默病(ad)、伴有路易体的痴呆和伴有帕金森病(pd)的额颞叶痴呆(ftd)之间的临床、病理和遗传重叠。在各种生化和细胞研究中,异常的硫酸乙酰肝素(hs)代谢正在成为ad和pd的常见致病机制,然而,潜在的机制尚未清楚。硫酸乙酰肝素蛋白聚糖(hspg)调节细胞培养物中淀粉样蛋白(aβ)和α
‑
突触核蛋白(α
‑
syn)的寡聚化、清除、内吞作用和转运。hspg与aβ结合,并且加速其寡聚化和聚集。hs在体外独立地刺激α
‑
syn原纤维的形成,并且介导细胞aβ摄取。hspg还介导α
‑
syn的巨嗜细胞摄取。药理学上抑制hspg与致病蛋白的结合以及遗传上减少hspg合成有助于致病蛋白的清除并减少其聚集。此外,hspg存在于aβ斑块和路易体(lb)中。这些发现表明,hs和hspg在aβ和α
‑
syn代谢以及随后的ad和pd的发病机制中起重要作用。然而,尚未系统评估参与hs的溶酶体降解的酶中的遗传变体在ad或pd的发病机制中所发挥的作用。因此,在大病例
‑
对照ad和pd组群中进行了遗传分析,并在负责硫酸乙酰肝素(hs)降解的溶酶体酶基因中鉴定了罕见功能变体的显著富集,具有显著的效应大小。研究最广泛的参与hs降解的溶酶体酶是n
‑
乙酰基
‑
α
‑
氨基葡萄糖苷酶(naglu),其在人体内的缺乏会导致粘多糖贮积症iiib(mps
‑
iiib)。存在一种良好表征的naglu缺陷小鼠模型,该模型概括了人类疾病的特征。本文所述研究的目的是确定在ad(优势比=3.7)和pd(优势比=4.7)患者中发现的naglu基因的亚形错义变体是否影响app代谢、体外aβ生成和aβ降解、体内ad病理以及体外α
‑
syn聚集和体内α
‑
syn扩散。
[0556]
(i)确定naglu基因中的亚形错义变体对神经元app代谢和aβ生成、体外aβ胶质细胞降解和体内ad病理的影响
[0557]
人mps
‑
iiib患者和naglu缺陷小鼠表现出细胞内全长app的升高的皮质水平。与对照脑相比,mps
‑
iiib患者还表现出可溶性aβ40水平的显著的三倍升高。假设naglu活性的变化会影响app代谢和神经元中aβ的生成。携带naglu变体的ad患者的杂合状态在体外模拟。如本文所述,可以在原代神经元中确定亚形naglu变体对app转运、app半衰期、app加工机制和aβ生成的影响。
[0558]
越来越多的证据表明,小胶质细胞有助于ad的发病机制。数据挖掘结果显示,naglu在小胶质细胞中表现出高水平的表达。然而,关于naglu在神经胶质细胞中的作用知之甚少。因此,如本文所述,可以确定稳定地表达亚形naglu变体的原代小胶质细胞是否可以摄取和降解外源性aβ。
[0559]
在没有家族性ad突变的情况下,naglu缺陷小鼠在内侧内嗅皮层中表现出aβ和hspg的细胞内积累。在体内模拟了naglu杂合性和衰老对ad病理的影响。使用aav2/9
‑
php.b载体在出生时注射naglu半合子小鼠后,可以确定最有害的naglu变体是否影响24个月大的naglu半合子小鼠的ad相关表型。
[0560]
可以确定naglu减少或过表达是否影响5xfad小鼠在4个月和8个月时的aβ生成、aβ清除、斑块沉积、突触丢失和神经炎症。
[0561]
(ii)确定naglu基因中的亚形错义变体对体外α
‑
syn聚集和体内α
‑
syn扩散的影响
[0562]
mps
‑
iiib患者表现出黑质(sn)的严重神经元缺失,和在颞叶皮质、海马和sn中的神经元中病理性磷酸化的α
‑
syn(psyn)的积累。假设naglu活性的降低会影响α
‑
syn的摄取、
清除,以及体外聚集和体内扩散。
[0563]
如本文所述,可以确定在稳定地表达亚形naglu变体的原代神经元中,α
‑
syn预制原纤维(pff)的结合、内化和聚集是否受到影响。
[0564]
还可以确定底物减少(染料木素)、重组酶替代或基因疗法是否能缓解对α
‑
syn pff的内化和聚集的影响。
[0565]
在野生型和表达突变型a53t人α
‑
syn的转基因小鼠中,α
‑
syn pff的纹状体内注射再现了细胞内lb病理的积累、sn神经元的选择性丢失和运动协调受损。在注射了aav2/9
‑
php.b载体的半合子或naglu缺陷小鼠中进行α
‑
syn pff的纹状体内接种,该载体在出生时表达最有害的naglu变体。对磷酸化的α
‑
syn的聚集体的形成、连通性依赖性扩散及其对疾病进展和寿命的影响进行了量化。
[0566]
意义
[0567]
在ad和pd中的硫酸乙酰肝素
[0568]
在各种生化和细胞研究中,异常的硫酸乙酰肝素(hs)代谢正在成为ad和pd的常见致病机制,然而,潜在的机制尚不清楚。由共价连接到特定蛋白核心的hs链组成的硫酸乙酰肝素蛋白聚糖(hspg)是大量的细胞表面和细胞外分子,其与一系列配体相互作用。膜hspg充当内吞受体,并且经历组成型和配体诱导的内吞作用。大多数hspg和结合的配体被溶酶体蛋白酶、外糖苷酶和硫酸酯酶降解。hspg调控多种致病蛋白(包括aβ和α
‑
syn)的寡聚化、清除、内吞作用和转运。hspg存在于aβ斑块以及lb和ln中。显示,hspg与aβ结合,并且加速其寡聚化和聚集。在体外,hs显著地刺激α
‑
syn原纤维的形成。hs还介导细胞aβ的摄取。hspg介导α
‑
syn的大胞饮摄取。对致病蛋白的hspg结合的药理学抑制和对hspg合成的遗传性减少促进了致病蛋白的清除并减少了它们的聚集。这些发现表明,hs和hspg在aβ和α
‑
syn代谢以及ad和pd的发病机制中发挥重要作用。
[0569]
ad中的溶酶体功能障碍
[0570]
虽然家族形式的ad的病因是由增加的淀粉样β(aβ)生成以及随后aβ在细胞外间隙(isf,间质液)中聚集成可溶性低聚物或不溶性aβ斑块驱动的,但对晚发型散发性ad患者的最近研究表明aβ的受损的清除。因此,产生与清除之间的平衡决定了aβ水平和形成aβ斑块的倾向。溶酶体在细胞内细胞器和聚集倾向蛋白的降解中起主要作用。神经病理学研究还发现,ad脑中的溶酶体病理有助于ad的发病机制,然而潜在的机制尚不清楚。已在多种转基因小鼠ad模型中发现溶酶体的变化。这些结果一起表明,溶酶体蛋白的变化加速了ad的病理。细胞研究表明,内体
‑
溶酶体系统是aβ产生的主要场所。早老素2(psen2)和呆蛋白(催化必需的γ分泌酶组分)位于溶酶体中。事实上,psen1调节溶酶体ph。体外溶酶体功能的药理学损伤导致aβ产生的变化。溶酶体ph的变化会减少aβ的分泌。溶酶体蛋白酶抑制剂减少淀粉样变性app片段的产生。所有这些研究表明,整体溶酶体功能在正常和异常的aβ前体蛋白(app)加工及随后的淀粉样变性中起重要作用。
[0571]
人类和hs代谢基因缺陷小鼠中的app代谢
[0572]
在溶酶体中至少存在四种参与hs的分步分解的酶。这些基因中的功能丧失(lof)突变导致部分降解的hs在溶酶体中积累,并引起粘多糖贮积症(mps)iii型a、b、c和d(sanfilippo综合征)。mps患者在整个大脑的细胞的细胞质中表现出强烈的弥漫性aβ信号。与正常对照脑相比,mps患者表现出可溶性aβ水平的显著升高。此外,在没有家族性ad(fad)
突变的过表达的情况下,在mps小鼠模型中报告了细胞内app/aβ水平的升高。在mps iii小鼠模型中,发现aβ40水平比对照增加了三倍。人类和ad转基因小鼠的认知能力下降与可溶性aβ物种相关。来自转基因ad小鼠的数据表明,神经内aβ的神经毒性大于细胞外aβ。在人类和小鼠ad模型中,细胞内aβ的积累已显示先于细胞外沉积。这些研究表明,app转运或加工在人类患者和小鼠模型中都受到影响,其中参与hs代谢的溶酶体基因完全功能丧失。
[0573]
pd中的溶酶体功能障碍
[0574]
人类尸检研究和模型系统表明,内吞转运、溶酶体完整性和溶酶体水解酶活性的缺陷在突触核蛋白病中起重要作用。溶酶体标记物是患有散发性pd的患者中lb的组成部分。因此,已经建议,随着疾病的进展,lb和路易神经突(ln)可能在受损的溶酶体周围播散,并通过溶酶体来源的未降解材料的持续沉积而增大尺寸。多种基于细胞的模型集中于蛋白病变种子的细胞间转移在突触核蛋白病的进展中的中心地位,尽管机制问题仍然存在。尚不清楚特异性α
‑
syn菌株是否经由不同的受体或内吞机制被内化。通过永生化细胞和原代神经元对α
‑
syn的大胞饮摄取由hspg介导。然而,尚未评估hs在体内α
‑
syn扩散中的作用。溶酶体加工是原代神经元中内化的α
‑
syn原纤维的主要命运。溶酶体功能的药理学扰动导致α
‑
syn原纤维的细胞内加工的异常,伴随着经由募集内源性α
‑
syn形成包涵体的速率增加。对控制这种募集的过程仍知之甚少,并且这表明致病物种必须逃脱溶酶体内转运。因此,据报道,外源性α
‑
syn物种会导致内吞囊泡和溶酶体膜破裂,从而逃脱内吞转运和溶酶体降解。一旦进入胞质溶胶,这些α
‑
syn原纤维或寡聚体可以与可溶性物质相互作用,并启动内源性α
‑
syn的募集。这些结果进一步支持了溶酶体活性和完整性的缺陷可能加速病理性α
‑
syn聚集和传播的观点。hspg存在于lb和ln中。在体外,hs显著地刺激α
‑
syn原纤维的形成。然而,尚未完全确定hs的溶酶体积累在α
‑
syn聚集和扩散中的作用。
[0575]
人溶酶体基因中的遗传变异的大规模分析
[0576]
最近对60,000个人的外显子组中存在的突变的频率和类型的分析发现,溶酶体基因对功能丧失突变“不耐受”,并且携带的潜在有害变体比由中性进化模型预测的要少。这与敲除小鼠中大多数“核心”溶酶体基因在胚胎发生或新生儿期的致死性一致。研究最多且最广为人知的溶酶体基因是那些在突变时导致溶酶体贮积病(lsd)的基因。有趣的是,这些引起lsd的基因中的大多数对lof突变不耐受,并且在人类中表现出相当大的遗传编码变异。因此,在健康人中的溶酶体酶活性水平存在十倍范围的差异。长期以来,人们一直认为携带溶酶体基因的一个单一正常拷贝不会对健康造成影响。然而,最近对npc1基因中的杂合性错义致病突变的携带者的研究揭示了显著的全身代谢异常。此外,大量的遗传证据支持了gba基因中的杂合错义变体作为发展突触核蛋白病的主要遗传风险因素的作用。纯合状态下导致儿童lsd(戈谢病)的相同致病变体(当以杂合方式存在时)会影响成人发作的神经退行性疾病(包括pd和路易体痴呆)的风险。这些研究表明,衰老和溶酶体基因中的单倍体不足性的组合容易导致成人中常见的神经退行性障碍。缺乏对影响ad或pd风险的溶酶体基因中功能性杂合变体的贡献的系统评估。
[0577]
携带导致naglu基因中的单倍体不足性的遗传变体的个体表现出明显低于对照的酶活性水平。最近的研究报告了exac数据集中164种naglu错义“未知意义的变体(vus)”的酶活性,以及在hgmd中报告的35种致病性错义突变,其中17种也在exac中发现。表明,与野生型水平相比,大约90%的致病变体表现出<15%的酶活性。这些数据表明,在一般群体中
存在携带naglu基因中的亚形杂合变体的个体。然而,尚未完全研究naglu单倍体不足性的长期后果。在大病例
‑
对照ad和pd组群中进行了遗传分析,并且鉴定了溶酶体硫酸乙酰肝素(hs)代谢酶的基因中的罕见功能变体的显著富集,具有显著的效应大小。发现naglu中的亚形错义变体与ad(p=3
×
10
‑3;or=2.3;95%ci 1.2
‑
5.2)和pd(p=3.6x10
‑7;or=3.6;ci=2.8
‑
8.3)相关联。
[0578]
在ad或pd患者中发现的naglu基因中的亚形错义变体会影响app代谢、体外aβ生成和aβ降解,和体内ad病理以及体外α
‑
syn聚集和体内α
‑
syn扩散。
[0579]
创新
[0580]
本文所述的研究通过系统和全面地评估与ad和pd风险相关联的naglu基因中的亚形杂合变体的功能后果而在概念上是创新性的。来自这些研究的结果对具有遗传决定的溶酶体功能障碍的pd和ad患者的鉴定具有重要意义,其中此类功能障碍的恢复可以提供有效的疗法。本文还描述了利用底物减少、重组酶替代或基因疗法的挽救实验,这些实验可能具有翻译含义。
[0581]
一个将计算方法和实验数据相结合的创新性综合框架被用于验证naglu基因在体外和体内的功能效应。本文所述的该研究结合了最先进的技术和来自大数据集中的遗传分析的结果、基于细胞的测定、生化数据、来自小鼠和人脑中特定细胞类型的rnaseq数据、人类ad和pd病例和对照中的rna
‑
seq数据,以及来自ad小鼠模型的全基因组基因表达数据,这些数据与采用干预措施(诸如基因疗法、底物减少和重组酶替代)的小鼠模型中的aβ斑块、生化、组织病理学和行为数据相关。
[0582]
第(i)和(ii)节中描述的研究解决了年龄和naglu在易损脑区上的单倍体不足性是否影响app加工和转运、aβ斑块负荷、aβ水平和体外α
‑
syn聚集以及体内α
‑
syn扩散的问题。还在ad和pd机制中评估了naglu和细胞自主性(神经元相对于神经胶质细胞)的作用。
[0583]
本文描述了一种在小鼠中对与人神经退行性疾病相关的遗传变体进行建模的创新方法。在半合子小鼠的生命早期(出生后第2
‑
4天)注射嗜神经aav载体,该载体能够实现携带亚形变体的目的基因的非侵入性(通过脑血屏障)、广泛分布和持久的整体神经表达,并进行长达20
‑
24个月的长期随访。因此,不仅研究了已鉴定的遗传变体的细胞效应,而且还研究了衰老和杂合性的影响,如在ad和pd患者中所发现的。
[0584]
一个高效和跨学科的研究团队已经被集合起来执行该项目。这些研究只有通过合作创新才是可能的,所述合作创新涉及具有跨越神经遗传学、溶酶体生物学、lsd动物模型以及细胞和小鼠模型中ad和pd病理生理学的专业知识的研究人员。
[0585]
方法
[0586]
这里,使用最先进的功能基因组学工具来确定与ad和pd风险相关联的naglu遗传变异的体外和体内影响。
[0587]
研究
[0588]
溶酶体hs降解基因中的杂合变体影响发展ad的风险
[0589]
在两个病例对照的ad组群中对45个溶酶体基因进行了单变体和基于基因的分析。发现样品由来自667例不相关ad病例和511名对照的wes数据组成。如所预期的,来自exac数据集(欧洲人,非芬兰裔)的基因特异性积累等位基因频率(cmaf)与来自内部ad数据库的cmaf高度一致(r2=0.96)。将罕见蛋白改变变体(cmaf)的负荷与在对照和exac中观察到的
负荷进行比较。对于大多数基因,与对照相比,在病例中存在过多的变异,但仅发现与sgsh基因的名义关联(p=4.2
×
10
‑3;or=3.7;95%ci1.4
‑
9.6)。当与exac样品的cmaf相比时,sgsh基因(p=7.9
×
10
‑5;or=3.0;95%ci1.8
‑
4.9)和naglu基因(p=4.8
×
10
‑4;or=3.7,95%ci 1.4
‑
9.6)通过多重检验校正阈值p<1.0
×
10
‑3。接下来,使用阿尔茨海默病测序项目(adsp)组群(5045例ad病例和4500名对照)来重复这些发现。在该独立样品中复制了naglu(p=3
×
10
‑3;or=2.3;95%ci 1.2
‑
5.2)。值得注意的是以下事实,在复制样品中发现的关联在相同的方向并且具有相似的效应大小。
[0590]
随年龄、ad状态和ad小鼠模型中的naglu转录水平
[0591]
在神经病理学正常的人脑样品中,存在naglu转录水平随着年龄的显著增加(p=0.02)(参见,例如图1a)。与年龄匹配的对照相比,在ad病例中naglu转录水平显著较高(p=0.007,参见,例如图1b)。在两个较大且独立的ad数据库(梅奥诊所脑库rna
‑
seq和西奈山脑库(mayo clinic brain bank rna
‑
seq and mount sinai brain bank))中复制了ad患者中naglu转录水平的统计学意义(p=3.2x10
‑8)的升高(0.46log2倍)。与野生型小鼠中的水平相比(上图中的黑线,参见,例如图1c),在ad小鼠模型的皮质中,naglu转录水平也随着ad病理的发展(下图,参见,例如图1c)表现出成比例的年龄依赖性增加(蓝线上图;参见,例如图1c)。anova分析显示年龄和基因型的显著的相互作用(p=0.0042)。年龄解释了11.4%的变异(p=0.0003),而基因型解释了42.3%(p<0.0001)。这些结果表明,对来自naglu聚集蛋白的代偿性反应可能是正常衰老过程的一部分。在ad模型中发现的异常升高表明,它们试图控制aβ的异常水平。
[0592]
因此,naglu的单倍体不足性可能会加剧老年小鼠中的ad相关表型。
[0593]
naglu对aβ水平的间质液(isf)水平的基因剂量效应。
[0594]
对同窝仔wt、半合子和naglu缺陷小鼠中aβ的isf水平进行的微透析定量(参见,例如图14a)显示,naglu对aβ的基线isf水平具有基因剂量效应(参见,例如图14b)。在半合子和naglu缺陷小鼠中也发现isf aβ水平升高60%,但在对200μm氯喹有反应的野生型小鼠中未发现(参见,例如图14c)。在青年(3个月)或老年(18个月)app/ps1小鼠模型中,该剂量的氯喹未改变aβ的isf水平(数据未显示)。这些发现支持了naglu活性水平的变化与异常app代谢和aβ生成相关的假设。
[0595]
溶酶体hs降解基因中的杂合变体影响发展pd的风险
[0596]
发现样品由来自ppmi组群的331例不相关pd病例的wes数据组成。来自nfe exac数据集的基因特异性cmaf与来自内部pd数据库的cmaf高度一致(ppmi r2=0.92;wustl r2=0.96)。将罕见蛋白改变变体(cmaf)的负荷与在对照和exac中观察到的负荷进行比较。当与exac样品的cmaf比较时,七个基因通过多重检验校正阈值p<1.0
×
10
‑3),包括gba(p=6.1x10
‑6;or=2.1;ci=1.3
‑
3.3)、gns(p=2.5x10
‑5;or=2.4;ci=1.4
‑
4.6)和naglu(p=1.0x10
‑4;or=4.7;ci=1.5
‑
8.3)。在hgsnat中也存在趋势(p=8.1x10
‑3;or=1.8;ci=1.1
‑
2.8)。接下来,使用另一个pd组群(wustl)重复这些发现,包括490例pd病例,其中使用人类外显子组芯片获得数据。值得注意的是,在复制样品中发现的关联在相同的方向并且效应大小相似;naglu(p=3.6x10
‑7;or=3.6;ci=2.8
‑
8.3)和hgsnat(p=9.7x10
‑4;or=1.9;ci=1.4
‑
3.2)。在具有有naglu基因突变的mps iiib患者中,曾报告了sn病理和lb积累。此外,发现与对照相比,在来自pd患者的黑质的多巴胺能神经元中存在naglu基因的转
录水平的降低(参见,例如图2)。
[0597]
α
‑
syn的结合、内化和聚集的体外模型
[0598]
在神经元培养中,α
‑
syn pff的制备和使用已被优化。在体外(div)第7天,将α
‑
syn pff添加到来自野生型小鼠的原代皮质神经元中。治疗后7天;神经元被固定并用psyn特异性抗体染色。pff诱导内源性表达的α
‑
syn募集为异常的、磷酸化的、不溶性的聚集体(参见,例如图3a和图3b)。α
‑
syn聚集体最初在突触前终末和轴突中表现为小的点状内含物(参见,例如图3a)。聚集体生长,并且外观变得更细长和蜿蜒,类似于路易神经突(参见,例如图3a)。图3b显示,pbs处理的对照神经元显示出略高于15kda的条带,其对应于单体α
‑
syn。在用pff处理的神经元中出现了几条具有较高分子量的条带。那些额外的条带可能对应于α
‑
syn寡聚体。这是研究溶酶体功能障碍对α
‑
syn聚集的影响的易于处理的体外系统。
[0599]
psyn病理在体内的传播
[0600]
在六只naglu缺陷小鼠和六只野生型同窝小鼠中进行了α
‑
syn pff的纹状体内接种。所有小鼠均在注射后存活。与已发布的数据一致,注射后90天(dpi),pff注射的野生型小鼠在多个脑区(包括纹状体、额叶和内嗅皮质、基底外侧杏仁核和sn)中表现出丰富的同侧psyn病理。wt治疗的小鼠在内嗅皮层中表现出很少的对侧psyn病理(参见,例如图15)。用α
‑
syn pff治疗的naglu缺陷小鼠在注射部位的同侧和对侧的额叶和内嗅皮质中表现出更多的α
‑
syn病理(参见,例如图16b)。naglu缺陷小鼠似乎具有更多的病理学的psyn病理,这可能表明向同侧和对侧的扩散比在wt小鼠中观察到的要多。目前正在分析更多经α
‑
syn pff治疗的小鼠,以进一步表征naglu缺陷对psyn病理(尤其是sn中的psyn病理)的区域和时间扩散的影响(参见,例如图17a至图17c),并扩展了提示在naglu缺陷小鼠中psyn病理的扩散增加的初步结果。
[0601]
研究设计与方法
[0602]
(i)(a)在体外确定naglu基因中的亚形错义变体对神经元app代谢和aβ生成以及aβ胶质降解的影响
[0603]
naglu基因中的亚形错义变体
[0604]
使用定点诱变对所选的变体进行工程改造,并将所选的变体亚克隆到如前所述的慢病毒载体中。慢病毒载体按照涉及重组或合成核酸分子的nih研究指南的第iii
‑
e
‑
1节在bsl2设施中生产、处理和处置。为了确定变体对酶活性的影响,如前所述使用荧光测定法用于naglu活性。通过elisa对hspg的水平进行定量。
[0605]
评估对app转运、内吞作用和亚细胞定位的影响
[0606]
多项研究已经表明,app的内吞作用对于其在app淀粉样变性途径中与内体和多囊体内的β分泌酶和γ分泌酶共同定位是至关重要的。继发于溶酶体功能障碍的内体通量的损伤导致在该细胞器内的增加的转运时间,这增加了β裂解和γ裂解的倾向,并且因此增加了aβ的生成。据报道,在没有aβ斑块的情况下,naglu缺陷和sanfilippo b患者的大脑中细胞内全长
‑
app水平增加。为了确定naglu基因中的选定变体是否影响app的稳态水平、app内吞作用或app进入溶酶体用于降解的增强的通量,可以使用如先前公布的细胞表面生物素化测定法来确定细胞内app出现的动力学和细胞表面中app的水平。在用蛋白合成抑制剂环己酰亚胺处理0、5、10、30分钟时,通过蛋白免疫印迹来测量对全长app半衰期的影响。使用共定位技术以研究对app和sorl1亚细胞定位的影响。分别通过蛋白免疫印迹和rt
‑
qpcr来
测量app加工机制的蛋白和转录水平,包括α分泌酶(adam10和adam17)、β分泌酶1(bace1)和γ分泌酶复合物(psen1和呆蛋白)。为了确保严谨性和再现性,在至少3个独立实验中由双盲观察者一式三份进行定量。
[0607]
评估对aβ生成的影响
[0608]
很大一部分app被靶向溶酶体,并且在存在溶酶体酸化抑制剂的情况下,app水平在细胞中迅速升高,这表明溶酶体降解会驱动app蛋白水解,以阻止aβ肽的形成。与正常对照脑相比,sanfilippo b患者呈现出可溶性aβ40水平的显著升高(3倍)。据报告,在naglu缺陷小鼠的脑中,aβ寡聚体水平显著升高。这些发现表明,在具有naglu缺陷的小鼠和人类中,hs和溶酶体功能障碍的积累导致γ分泌酶依赖性异常app加工。因此,可以评估所选的变体是否影响细胞培养物中aβ的生成。如先前所述进行原代神经元培养。在神经元特异性启动子(突触素)下携带选定变体和野生型的慢病毒载体用于转导来自naglu缺陷和半合子小鼠的原代神经元。使用不同体积的浓缩慢病毒来确定神经元中的适当表达水平。通过rt
‑
qpcr和荧光测定法来测量naglu水平。通过如先前公布的夹心elisa来检测细胞裂解物和细胞培养基中的aβ物种。简而言之,用小鼠单克隆包被抗体hj2(抗aβ35
‑
40)和hj7.4(抗aβ37
‑
42)捕获aβx
‑
40和aβx
‑
42肽。hj5.1(抗aβ13
‑
28)(一种靶向中心结构域的生物素化抗体)或hj3.5(其靶向n
‑
末端氨基酸)被用作检测抗体,然后是链霉亲和素
‑
聚
‑
hrp
‑
40。通过蛋白免疫印迹来测量app衍生的蛋白水解片段,诸如α
‑
ctf和β
‑
ctf以及sappα和sappβ。如前所述,通过蛋白免疫印迹来监测全长app的水平。
[0609]
评估对aβ降解的影响
[0610]
小胶质细胞在aβ斑块周围增殖,并且吞噬aβ材料,但随后的降解受损,导致ad中进行性aβ积累。小胶质细胞为何可以摄取原纤维aβ但不能降解原纤维aβ尚不清楚。然而,来自ad患者的小胶质细胞显示beclin
‑
1的减少和随后的溶酶体功能障碍。此外,不溶性原纤维aβ影响氯离子通道cic
‑
7向原发性小胶质细胞中的溶酶体的转运,这损害溶酶体降解。然而,恢复溶酶体酸化会增强aβ的降解。总之,该证据表明小胶质细胞中的溶酶体不足性可能有助于ad发病机制。这里描述的数据挖掘工作揭示了,naglu在小胶质细胞中呈现出较高的表达水平。因此,可以评估来自用选定的亚形naglu变体转导的naglu缺陷和半合子小鼠的原代小胶质细胞是否可以摄取和降解外源性aβ。aβ的摄取和降解评估如先前公布的进行。
[0611]
评估对alp功能的影响
[0612]
在来自naglu缺陷小鼠的心脏和脑组织中增加的beclin1、p62和lc3
‑
ii水平表明溶酶体自噬系统的异常活性,伴有自噬体的积累。可以评估来自半合子naglu小鼠的神经元是否表现出alp功能障碍,以及这些变化是否通过所选的变体而增加。lc3和p62的蛋白免疫印迹可以用作巨噬细胞活化的间接指标。在不存在或存在自噬系统的激活剂(雷帕霉素和torin1)、自噬抑制剂(巴弗洛霉素a1)、溶酶体营养剂(氯喹、氯化铵)和e64/亮抑蛋白酶肽(如前所公布的)的情况下,通过细胞中存在的lc3
‑
ii的量来评估自噬通量。通过使用mcherry
‑
gfp
‑
lc3标记物的活细胞成像对此进行补充。可以通过lc3和lamp1的共定位来进一步评估自噬体
‑
溶酶体融合。溶酶体跟踪器用于定量每个细胞的酸性隔室的数目。tfeb的活化通过其核定位来评估。rt
‑
qpcr用于测量tfeb调节的mrna转录物(sqstm1/p62、map1lc3b和lamp2)的转录水平的变化。
[0613]
为了确保严谨性和再现性,研究者在定量和分析阶段期间对基因型视而不见。对
于每个实验,在所述的每个组中对获得的数据进行平均。实验以一式三份进行,每个基因型至少使用两种独立产生的制剂。使用双向anova和适当的事后检验来检验具有统计学意义的差异,以确定每种标记物或功能分析是否与naglu相对于对照相关联。
[0614]
预期的结果
[0615]
预期naglu对app转运、app代谢、aβ生成或aβ降解的基因剂量效应。此外,预期本文描述的实验允许评估naglu基因中选定的变体对神经元和小胶质细胞存活的影响。可替代地,可以用naglu基因中的选定变体对来自5xfad转基因小鼠的原代神经元或n2a695细胞进行转导,并且评估对aβ生成的影响。在体外鉴定出影响ad的风险和发病机制的naglu基因中的变体后,下一步是利用ipsc的生成和基因组编辑方法的进展。来自携带naglu基因中的变体的ad患者的ipsc衍生的神经元或胶质细胞可以用于比较此类变体与等基因crispr校正细胞相比对app代谢的影响。结合来自这里描述的实验的结果和naglu活性的荧光测定法的可用性,可以筛选ad病例和对照的csf、血浆、血清或脑组织,以检测可以用作ad的生物标记物的特定缺陷。
[0616]
(i)(b)在体内确定naglu基因对ad病理的影响
[0617]
虽然已经表征了naglu在小鼠和人类中完全功能丧失的神经退行性后果,但对该基因的单个拷贝的长期后果(单倍体不足性)或其过表达的了解非常少。一直以来,人们都认为半合子小鼠和人类是正常的。然而,最近表明,溶酶体基因中的单倍体不足性会导致在人类和小鼠中的显著的代谢异常。一个假设是ad病理由较温和形式的遗传性溶酶体功能障碍发展而来,并且其出现可能需要额外的年龄相关溶酶体损伤。主要终点为在4个月大时(在斑块沉积之前)测得的aβ水平和在8个月大时在小鼠中存在引起fad的突变的斑块负荷。对aβ水平的影响是表达与ad相关联的最有害naglu变体的24月龄naglu半合子小鼠中的主要终点。
[0618]
亚形错义naglu变体对老年小鼠ad表型的影响
[0619]
对同窝仔wt、半合子和naglu缺陷小鼠中aβ的isf水平进行的微透析定量(参见,例如图14a)显示,naglu对aβ的基线isf水平具有基因剂量效应(参见,例如图14b)。在半合子和naglu缺陷小鼠中也发现isf aβ水平升高60%,但在对氯喹有反应的野生型小鼠中未发现(参见,例如图14c)。ad病理(例如,aβ斑块)通常具有年龄依赖性。然而,已发布的研究尚未解决年龄与溶酶体功能障碍之间的相互作用。大多数评估体内作用的研究已经采用了药理学方法,或完全没有溶酶体基因和短期终点。以前曾报告在10个月大的naglu缺陷小鼠的大脑中出现aβ寡聚体的增加。这里,在出生后第1
‑
2天通过i.v向半合子naglu小鼠注射aav2/9
‑
php.b,其携带最有害的naglu变体。aav2/9
‑
php.b载体通过血脑屏障,并且以比aav9高至少40倍的效率在整个cns中转移基因,并转导多个cns区域中的大多数神经元。允许小鼠恢复并存活至至少20个月大。将垂死的小鼠麻醉并处死,以收集脑生化研究结果,诸如洗涤剂可溶性和不溶性aβ40和aβ42水平。还对涉及aβ的ad相关表型进行了定量病理学调查(参见,例如图18)。
[0620]
样品量计算表明,至少需要n=15只小鼠/组来检测具有80%功效的洗涤剂可溶性和不溶性aβ40和aβ42的20%增加。在出生后第1
‑
2天使用aav2/9
‑
php.b向十五(15)只naglu半合子小鼠和15只野生型小鼠注射最有害的naglu变体。
[0621]
naglu减少或过表达对小鼠ad病理模型的影响
[0622]
在患有sanfilippo b病的人类患者中,naglu蛋白的完全功能丧失导致可溶性aβ40的水平比正常对照脑显著升高三倍。然而,在人类ad病例和ad小鼠模型中,naglu转录水平显著高于对照(参见,例如图1b和图1c)。考虑到来自人sanfilippo b患者和naglu缺陷小鼠的数据支持这些基因在细胞内aβ生成中的作用,在良好表征的携带有利于细胞内aβ生成的fad突变的ad小鼠模型中,可以确定naglu减少或过表达是否影响aβ生成。5xfad模型是一种aβ沉积模型,其在1.5个月时发展为神经内aβ42,在2个月时发展为斑块,在4个月时发展为突触标记物的丧失和记忆缺陷,并且在9个月时发展为神经元丧失。斑块的发展伴有反应性胶质增生。naglu缺陷小鼠表现出高度硫酸化的hs脑积累、神经炎症、神经元和小胶质细胞中的溶酶体增大以及约4mo的突触蛋白减少,随后是昼夜节律改变、听力和视力缺陷,以及在老年小鼠(>8mo)中,浦肯野细胞的丧失和运动协调受损。naglu缺陷小鼠的中位寿命为约12月龄。naglu和5xfad小鼠在c57bl/6背景下是同源的。为了进一步确定naglu单倍体不足性是否会加剧现有的淀粉样变性过程,将naglu小鼠与5xfad转基因小鼠杂交。可以确定naglu的过表达是否会影响5xfad小鼠中的ad病理。在5xfad小鼠出生时,使用携带野生型naglu的aav2/9进行单侧单次颅内注射。然后,如前所述,通过组织学测量aβ斑块负荷,并通过夹心elisa在第4和第8个月测量aβ40/aβ42水平,在每只5xfad小鼠的aav2/9
‑
wt
‑
naglu注射的半脑和未注射的半脑中比较aβ斑块负荷。通过经由蛋白免疫印迹测量app
‑
ctf来评估app代谢。在5xfad小鼠中,四个月是aβ斑块沉积的早期时间点,以便检测aβ积累是否开始得更早,而八个月代表当aβ斑块丰富时的晚期。
[0623]
aav2/9载体的产生
[0624]
naglu野生型和最有害的变体被亚克隆到aav2/9
‑
php.b或aav2/9载体中。从unc病毒载体核心机构获得了高滴度的aav2/9载体储备液。对于本研究中概述的所有实验,在乳酸林格氏溶液中将aav载体储备液稀释至10
12
vg/ml。
[0625]
aβ斑块定量和aβ生成
[0626]
固定的冷冻脑切片(50μm)在小鼠的亚组群中用x
‑
34进行染色,并用hj3.4(抗aβ)抗体进行免疫染色,以量化斑块负荷(以%面积表示)。将来自对侧半球的脑组织匀浆中的aβ水平分级为可溶性(pbs)和不溶性(5m胍)级分,并使用elisa进行定量。评估了对app加工机制的影响。
[0627]
突触标记物
[0628]
突触丢失是人类和ad小鼠模型中的常见发现。可以通过蛋白免疫印迹(使用针对突触前标记物:snap
‑
25、囊泡相关联膜蛋白2、突触结合蛋白1和突触素的抗体,如前所述)来评估naglu的减少或过表达是否会影响5xfad小鼠中的突触丢失。
[0629]
体内aβ微透析
[0630]
aβ在大脑中具有相对短的半衰期,其中在小鼠间质液(isf)中为约1
‑
2小时并且在人脑脊液(csf)中为约8小时。为了研究naglu对aβ生成和清除的影响,使用体内微透析来动态地评估在3
‑
4个月大时注射avv2/9或pbs的5xfad/naglu(+/
‑
)小鼠、5xfad/naglu(
‑
/
‑
)小鼠和5xfad同窝小鼠的海马中的isf aβ代谢。为了评估清醒、自由活动小鼠的海马中isf aβ水平随时间的变化,如前所述进行了体内微透析(参见,例如图14a至图14c)。简而言之,在异氟醚麻醉下,立体定向地将引导套管植入到海马上方(前囟后3.1mm,中线旁2.5mm,和硬脑膜下1.2mm,呈12
°
角)。微透析探针通过引导套管被插入到大脑中。人工csf被用作微透析
003780)])。
[0643]
(ii)确定naglu基因中的亚形错义变体对体外α
‑
syn聚集和体内α
‑
syn扩散的影响
[0644]
如这里所述,可以在体外确定naglu对α
‑
突触核蛋白的摄取、转运、聚集和清除的功能作用。永生化细胞和原代神经元对α
‑
syn的大胞饮摄取似乎是由hspg介导的。此外,hs在体外显著地刺激α
‑
syn原纤维的形成。已经在培养的神经元中建立了良好表征的α
‑
syn聚集模型。在该模型中,将由重组α
‑
syn生成的pff直接添加到原代神经元中,并被神经元内吞。这些pff诱导内源性表达的α
‑
syn募集为异常的、磷酸化的、不溶性和泛素化的聚集体。体外来源于野生型的非转基因小鼠的原代神经元中由内源性α
‑
syn形成这些聚集体遵循2
‑
3天的滞后期,随后在轴突中在第4
‑
7天形成,在第7
‑
10天扩散至体细胞树突状隔室中,并在加入pff后约14天神经元死亡(参见,例如图3a至图3b)。如这里所述,使用这一良好表征的模型,可以确定naglu对α
‑
syn原纤维的摄取、转运、聚集和清除的影响。还可以确定在来自用最有害的naglu变体转导的naglu缺陷和半合子小鼠的原代海马神经元中内源性α
‑
syn的清除率是否存在变化。可以确定在用α
‑
syn pff治疗后(参见,例如图3a至图3b)在来自naglu缺陷小鼠的原代神经元中α
‑
syn聚集体(包涵体)的速率和水平。还可以确定底物减少(染料木素)、重组酶替代或基因疗法替代是否可以挽救来自naglu缺陷小鼠的神经元中的α
‑
syn pff。
[0645]
α
‑
syn pff的产生
[0646]
从细菌中制备重组单体α
‑
syn,并根据所建立的方案通过尺寸排阻和离子交换色谱法依次纯化。通过在37℃下搅动重组单体持续约72
‑
120小时,然后使用具有指定分子量截止参数的离心过滤装置进行尺寸选择,来制备原纤维形式的α
‑
syn。用于生成pff的条件先前已经被优化。pff在tris缓冲的nacl中稀释,并且在5
‑
10div后加入到培养的原代神经元中。暴露4
‑
7天后,通过免疫荧光或顺序提取和免疫印迹来确认pff转导和接种。通过用抗psyn(ser129)抗体、克隆81a(biolegend,mms
‑
5091)免疫荧光并且通过蛋白免疫印迹来检测来自内源性α
‑
syn的异常α
‑
syn聚集体。α
‑
syn pff在bsl2设施中生产、处理和处置。
[0647]
α
‑
syn pff转运、内吞作用和亚细胞定位
[0648]
溶酶体加工是原代神经元中内吞的α
‑
syn原纤维的主要命运。用突触前(cspα)、内吞(eea1)、自噬体(rab7)和溶酶体(lamp1)标记物对pff处理的神经元进行共染色,以确定在内溶酶体途径中α
‑
syn聚集体的转运是否存在变化。
[0649]
评估对alp功能的影响
[0650]
α
‑
syn聚集体通过降低自噬体清除率而损害整体巨噬细胞。因此,可以确定对自噬途径的药理学调节(如上所述)是否能改善α
‑
syn的清除率。
[0651]
评估对伴侣介导的自噬(cma)的影响
[0652]
α
‑
syn被伴侣介导的自噬(cma)降解,并且易于聚集的α
‑
syn突变体阻断cma。因此,可以确定lamp2a和hsp70蛋白水平在pff处理的细胞中是否受到影响。用cma激活剂处理pff处理的神经元;在细胞裂解液和条件培养基中确定ar7(视黄酸受体α特异性拮抗剂)和α
‑
syn水平。
[0653]
对内吞和溶酶体膜完整性的影响
[0654]
α
‑
syn pff诱导内吞后囊泡和溶酶体破裂。这些破裂的囊泡对eea1、lc3和半乳糖凝集素
‑
3呈阳性。通过gal
‑
3和lc3染色来确定α
‑
syn pff是否影响溶酶体膜的完整性。
[0655]
挽救实验
[0656]
在存在或不存在摄取/结合抑制剂(甘露糖
‑6‑
磷酸:m3655,sigma)的情况下,在暴露于α
‑
syn之前、期间和之后添加重组naglu,并检测对α
‑
syn pff聚集的影响。已经存在足够的重组naglu用于在体外添加到缺陷神经元中。染料木素(4',5,7
‑
三羟基异黄酮或5,7
‑
二羟基
‑3‑
(4
‑
羟基苯基)
‑
4h
‑1‑
苯并吡喃
‑4‑
酮)抑制mps患者的培养的成纤维细胞中糖胺聚糖(gag)的合成,并且减少naglu缺陷小鼠中的溶酶体贮积物质。染料木素用作体外实验的底物还原剂。
[0657]
预期的结果
[0658]
预期naglu在体外对α
‑
syn pff的摄取、转运和聚集具有基因剂量效应并且重组酶将挽救那些效应。naglu缺陷细胞在细胞膜和内溶酶体系统中积累hs和hspg。因此,预期α
‑
syn pff的摄取增加,随后内吞囊泡和溶酶体膜破裂,这将增加胞质水平和内源性α
‑
syn的募集。可以生成慢病毒载体,并且易于聚集的α
‑
syn突变体可以在来自naglu缺陷和半合子小鼠的原代神经元中过表达。然后评估聚集和降解的速率。可替代地,可以使用来自过表达人a53tα
‑
syn转基因小鼠的原代神经元,并且可以用naglu基因中的选定变体对它们进行转导,并且测试对α
‑
syn的影响。来自携带naglu基因中的变体的pd患者的ipsc源性神经元可以用于比较此类变体与等基因crispr校正的细胞相比对α
‑
syn加工的影响。
[0659]
确定naglu对体内α
‑
syn扩散的影响
[0660]
积累的实验数据表明,α
‑
syn的细胞间传递遵循与针对朊病毒蛋白观察到的接种原则类似的接种原则。向过表达人a53tα
‑
syn的年轻小鼠(约3
‑
4月龄)中脑内注射含有聚集的α
‑
syn的脑提取物(来自路易体病的病例的尸检脑提取物)会刺激在宿主中形成psyn损伤,早在注射后30天(dpi)被观察到;到约90dpi,psyn在脑的解剖连接区域中广泛且丰富,这表明与在人类pd病例中观察到的α
‑
syn沉积的明显扩散类似(参见,例如图15、图16a至图16b和图17a至图17c)。在100dpi左右,与未注射的小鼠相比,小鼠出现运动功能障碍和过早死亡(约126dpi)。在非转基因(野生型)宿主小鼠中,脑内注射合成(人或小鼠)α
‑
syn pff也诱导了lb样病理和神经元变性(参见,例如图15、图16a至图16b和图17a至图17c)。大约有50%的注射有具有lb脑的痴呆的不溶性psyn的野生型小鼠发展了psyn病理。相比之下,由人和小鼠α
‑
syn pff诱导psyn病理的效率分别为90%和100%。在30dpi时,psyn阳性的lb样积累仅在注射部位同侧(参见,例如图15、图16a至图16b和图17a至图17c)。在受影响的同侧区域内以及在对侧新皮质内的lb/ln病理显示在90和180dpi检查的小鼠中,psyn免疫反应性显著增加。在注射pff后,sn致密部(snpc)α
‑
syn病理逐渐发展,从30dpi时的淡细胞质积累演变为90和180dpi时的致密核周lb样包涵体,尤其是在腹内侧snpc群体中。snpc多巴胺能(da)神经元在90和180dpi时分别伴随减少15%和35%,表明lb/ln在snpc da神经元丢失之前形成。因此,lb/ln的传播是连接性依赖的,并且病理性α
‑
syn的积累似乎是在snpc da神经元丢失的上游并与snpc da神经元丢失直接相关。这种纹状体内注射α
‑
syn pff的体内模型概括了细胞内lb/ln病理的积累、snpc da神经元的选择性丢失和受损的运动协调性。lb和ln都包含hspg。然而,hs在体内α
‑
syn扩散中的作用尚未得到评估。到目前为止,还不清楚naglu活性的降低和所产生的hspg积累是否影响α
‑
syn的聚集和扩散。如本文所述,表达与pd相关联的最有害naglu变体的naglu缺陷和半合子小鼠可以用于测试hs和hspg积累是否影响α
‑
syn聚集、扩散和加速体内疾病。
[0661]
α
‑
syn pff的纹状体内接种
[0662]
如上所述来制备α
‑
syn pff。在出生时使用aav2/9
‑
php.b在注射了最有害的naglu变体的naglu缺陷和半合子小鼠(参见,例如图19)以及naglu缺陷小鼠、半合子小鼠和十二只野生型小鼠中,对α
‑
syn pff进行纹状体内接种。量化了psyn聚集体的扩散,并且确定了α
‑
syn pff是否会影响naglu小鼠的寿命。在单次接种小鼠αsyn pff(或作为对照的pbs或αsyn单体)的情况下,对3
‑
4月龄野生型、naglu缺陷或半合子小鼠在背侧纹状体(0.2mm a/p,相对于前囟2.0mm m/l,颅骨表面下3.2mm)中单侧注射。允许小鼠恢复并在注射后长到年龄至30、90或180天,此时收获大脑并用抗psyn(ser129)抗体进行免疫组织化学处理。使用抗泛素和hsp90进行psyn共定位染色。基于功效分析,需要n=12只小鼠/组以检测psyn水平的30%增加。(5组x 12=60只小鼠)。
[0663]
αsyn和psyn的定量
[0664]
从每只动物的半脑中解剖皮质、海马、纹状体和脑干,并且通过顺序去污剂提取来分离不溶性αsyn,然后使用抗突触核蛋白
‑
1/克隆42(bd biosciences)和抗psyn 81a(biolegend)作为捕获抗体进行elisa和蛋白免疫印迹(如前所公布的)。
[0665]
使用如先前公布的针对酪氨酸羟化酶(th)和多巴胺转运体(dat)的抗体,对来自pff治疗的动物和pbs治疗的动物的同侧和对侧纹状体进行免疫印迹分析(参见,例如图17c)。评估sn中的脑萎缩和神经元丢失,并如上所述进行损伤和炎症的标记物(例如gfap,iba
‑
1)的免疫组织化学。
[0666]
比较pff处理的动物与pbs处理的动物之间的寿命。已知在野生型小鼠中,α
‑
syn pff注射6个月后,旋转棒和悬丝测试对于检测运动障碍是最灵敏的。之前已经在lsd小鼠模型中公布了旋转棒和线悬挂行为测试的使用,并且已知适当的实验设计和统计工具(用于多组比较的具有事后分析的anova,以及用于成对比较的学生t检验)。在pff治疗的动物和pbs治疗的动物中进行步态分析。之前已经表明,在lsd的小鼠模型中,步态分析对捕捉“帕金森样”体征非常敏感。
[0667]
样品量
[0668]
关于旋转棒和悬丝测定的性能,对于一次五组之间的比较,当正常动物在60秒下进行并且naglu缺陷动物在0秒(40周)下进行,并且标准偏差为30秒时,效应大小为0.89。在0.89的效应大小,α=.05,和功效=.95的情况下,仅需要6只动物就能够检测到组之间的显著差异。关于寿命,对于中位寿命为约730天的正常动物和半合子动物与中位寿命为约322天的naglu缺陷动物且标准差为20天的五个组之间的比较,效应大小为5.51。在5.51的效应大小,α=.05和功效=.95的情况下,仅需要2
‑
3只动物就能够检测到组之间的显著差异。由于使用了10
‑
12只小鼠/组,因此行为和寿命研究具有足够的效力。
[0669]
预期的结果
[0670]
预期α
‑
syn pff注射将加速naglu缺陷小鼠的表型,增加胶质增生、神经变性,使运动障碍恶化和使寿命缩短。半合子naglu小鼠将使psyn病理的传播恶化。可替代地,可以生成携带α
‑
syn突变的aav2/9载体,并立体定向地注射到年轻naglu缺陷小鼠的纹状体中,或者可以将过表达的人a53tα
‑
syn小鼠杂交到naglu缺陷小鼠中,并观察psyn病理、疾病进展和寿命的变化。同样的方法可以应用于sgsh缺陷小鼠。
[0671]
实例5:鉴定参与帕金森病发病机制的自噬
‑
溶酶体途径(alp)的功能障碍背后的
遗传变异
[0672]
本实例描述了鉴定pd中富含罕见功能变体的alp基因,并确定特异性alp基因在体外对α
‑
突触核蛋白聚集体和线粒体自噬的功能作用。
[0673]
新出现的证据强调了帕金森病(pd)和某些溶酶体贮积症(lsd)之间重叠的临床、病理和遗传特征,表明了共同的致病机制。gba、smpd1和npc1基因中的纯合子功能丧失突变会导致lsd。gba、smpd1和npc1中的杂合低频编码变体会增加pd的风险。这些结果表明,alp基因中的完全功能丧失变体会导致lsd,并且部分功能丧失低频变体会增加pd的风险。然而,尚未对alp中各基因的遗传变异对发展pd风险的贡献及其在pd发病机制中的作用进行系统和全面的评估。为了解决现有知识中的这一空白,本文描述了一种强有力的方法,来鉴定并优先考虑alp(除gba外)的基因中与发展pd的风险相关联的罕见功能变体。首先,将来自33,350名非芬兰裔欧洲人的alp各基因中罕见功能变体的负荷与2,700例pd病例和2,000名对照进行比较。这里显示的结果表明,在包括gba、naglu和ppt1的pd患者中存在至少六个引起lsd的基因的预测功能变体的富集。其次,使用基于细胞的测定来检查与pd风险相关的naglu和ppt1基因中所选的变体对酶活性、蛋白稳定性和/或mrna水平的影响及其对总溶酶体功能的影响。在体外评估了经验证的功能变体对α
‑
突触核蛋白(α
‑
syn)聚集清除率和线粒体自噬的影响。在暴露于α
‑
syn预制原纤维(pff)后,还测量了naglu和ppt1基因的基因剂量对内源性α
‑
syn聚集体的影响。然后可以确定重组酶替代是否可以减轻来自naglu和ppt1缺陷小鼠的神经元中α
‑
syn预制原纤维的pff诱导的毒性。最后,可以确定naglu和ppt1缺陷小鼠的原代多巴胺能神经元中是否存在线粒体自噬的变化。这里,所述的研究可以对pd发病机制中alp功能障碍的机制提供更深入的了解,并且可以为将目前针对lsd的治疗策略重新用于pd的潜在治疗奠定基础。
[0674]
本文所述研究的目的是确定帕金森病(pd)发病机制中自噬
‑
溶酶体途径(alp)功能障碍背后的遗传变异。这里介绍的研究并入了整合框架,该整合框架将人类遗传研究与选定alp基因对α
‑
突触核蛋白聚集体和体外线粒体自噬的功能验证相结合。这里描述的实验可以揭示与pd相关联的新alp基因。它们可以对pd发病机制中alp功能障碍的机制提供更深入的了解,并且可以为将目前针对溶酶体贮积症的治疗策略重新用于pd的潜在治疗奠定基础。
[0675]
新出现的证据强调了pd与某些溶酶体贮积症(lsd)之间重叠的临床和遗传特征,表明可能存在共同的致病机制。多个孟德尔和pd风险相关联基因(包括gba、lrrk2、vps35、snca、gak、atp13a2和rab7l1)参与自噬
‑
溶酶体途径(alp),并且其变体促进alp功能障碍。gba、smpd1和npc1基因的纯合子功能丧失突变会导致lsd。有趣的是,gba、smpd1和npc1中的杂合低频编码变体会增加pd的风险。这些结果表明,alp基因中的完全功能丧失变体会导致lsd,而部分功能丧失的低频变体会增加pd的风险。对分离群体中单个alp基因的研究支持了pd与lsd之间的遗传重叠。然而,尚未在一般群体中对alp各基因的遗传变异对发展pd风险的贡献及其在pd发病机制中的作用进行系统和全面的评估。
[0676]
(i)鉴定pd中富含罕见功能变体的alp基因
[0677]
假设alp基因(除gba外)中未经测试的罕见功能变体会增加pd的风险。如这里所述,分析了来自33,350名对照(exac数据库,非芬兰欧洲人)的全外显子组测序(wes)数据,以确定欧洲血统(ea)的个体的alp(≈430)各基因中的基线遗传变异。接下来,在pd病例对
照组群(2,700例病例/2,000名对照)中,结合外显子组芯片、全基因组(gwa)和wes数据进行alp基因的基于基因的分析。先前已在这些pd组群中鉴定出六个引起lsd的基因中的预测罕见功能变体的富集。
[0678]
(ii)确定特异性alp基因对α
‑
突触核蛋白聚集体和体外线粒体自噬的功能作用
[0679]
患有婴儿神经元性蜡样脂褐质病或粘多糖贮积症iiib的患者表现为帕金森综合征、黑质变性或路易体积累。在pd患者的da神经元中存在n
‑
乙酰基
‑
α
‑
氨基葡萄糖苷酶(naglu)和棕榈酰蛋白硫酯酶1(ppt1)基因的转录水平的降低。与(i)中提供的数据一起,选择了naglu基因和ppt1基因,以进一步验证在体外参与pd发病机制。
[0680]
多项严格标准表明(i)中鉴定的罕见变体具有潜在功能。为了验证其功能效应,用携带3种变体的慢病毒载体转导来自naglu和ppt1缺陷小鼠的原代神经元,所述3种变体被预测为(i)中鉴定的每个基因的最有害变体,包括先前鉴定为致病的两种变体。测量了naglu基因和ppt1基因中所选的变体对酶活性、蛋白和rna水平的影响。还确定了对总体溶酶体功能的影响。
[0681]
多项体外和体内药理学研究表明,alp功能障碍会导致聚集的α
‑
突触核蛋白(α
‑
syn)的有缺陷的清除。然而,尚不清楚特异性alp基因(除gba外)的缺失是否会影响聚集的α
‑
syn的溶酶体清除率并增加其毒性。这里,描述了可以确定naglu和ppt1基因的缺失是否影响内源性α
‑
syn的清除率的研究。α
‑
syn在来自naglu和ppt1缺陷小鼠的原代神经元中被过表达,并测试其对溶酶体清除率和神经毒性的影响。还可以确定向来自naglu和ppt1缺陷小鼠的原代神经元中添加外源性α
‑
syn预制原纤维(pff)是否会加剧pff诱导的神经毒性。最后,可以确定重组酶替代是否可以减轻来自naglu和ppt1缺陷小鼠的神经元中α
‑
syn预制原纤维的毒性。生成和纯化α
‑
syn pff的条件已在之前被优化。野生型海马神经元也已经用α
‑
syn转导,并且使用荧光和蛋白免疫印迹方法检测到磷酸
‑
α
‑
syn包涵体。
[0682]
线粒体功能障碍有助于pd的发病机制。lsd的溶酶体功能障碍与线粒体质量控制途径的调节失调和受损的线粒体的积累相关联。线粒体自噬由引起pd的基因parkin和pink1介导。然而,尚不清楚线粒体自噬是否受naglu或ppt1基因缺乏的影响,以及是否与parkin/pink1途径相关。这里,描述了可以确定在来自naglu和ppt1缺陷小鼠的原代多巴胺能神经元中是否存在线粒体自噬变化的研究。
[0683]
意义
[0684]
在人类基因组中,至少有430个与自噬
‑
溶酶体途径(alp)相关联的基因(38个自噬基因、161个自噬调节基因、64个溶酶体基因和167个溶酶体调节基因)。所有alp基因中的38%(157个基因)的突变导致孟德尔病(omim),其中研究最多的是经典的溶酶体贮积症(lsd)。存在至少50种不同的lsd,其作为一个组,在5,000例活产中出现频率为约1。lsd通常被视为儿科障碍,并且通常由完全功能丧失(lof)突变引起。然而,已经报道了携带亚形变体的成人发作形式的lsd。lsd为单基因障碍,但可以表现出复杂的临床特征。事实上,大约75%的lsd具有临床意义的神经系统组分。
[0685]
新出现的证据揭示了pd与某些溶酶体贮积症(lsd)之间重叠的临床和遗传特征,表明存在共同的致病机制。gba、smpd1和npc1基因中的纯合子功能丧失突变会导致lsd。有趣的是,gba、smpd1和npc1中的杂合低频编码变体会增加pd的风险。这些结果表明,alp基因中的完全功能丧失变体会导致lsd,而部分功能丧失的低频变体会增加pd的风险。对分离群
体中单个alp基因的研究支持了pd与lsd之间的遗传重叠。然而,尚未在一般群体中对alp各基因中的遗传变异对发展pd风险的贡献及其在pd发病机制中的作用进行系统和全面的评估。
[0686]
新生儿筛查研究显示,在健康人体内,溶酶体酶活性水平的范围超过十倍。gba、npc1、galc、gaa、gla和idua基因中致病变体的杂合携带者表现出比对照显著更低的酶活性水平。此外,npc1中致病变体的杂合携带者在受尼曼
‑
匹克影响的主要途径下游表现出显著的代谢异常。迄今为止,不存在关于此类代谢改变和alp功能障碍的长期后果的系统研究。然而,多项研究表明,致病变体的杂合携带者具有较高的神经退行性疾病风险。这里描述的研究解决了当前知识中的多个空白。首先,使用来自大型数据库中的全外显子组测序(wes)数据来确定欧洲血统(ea)的个体的alp的每个基因中的基线遗传变异。其次,分析pd病例中alp的每个基因中罕见功能变体的积累等位基因频率,以确定影响发展pd的风险的新基因。最后,体外验证了这些候选基因在pd的发病机制中的作用。通过鉴定pd中alp基因的特定缺陷,可以利用目前治疗lsd的多种治疗策略,包括基因疗法、酶替代、口服小分子底物减少疗法、小分子伴侣和自噬途径的药理学补救,以用于pd的潜在治疗。这些数据将为更彻底的实验奠定基础,这些实验被设计成确定这些alp基因在体内的作用。
[0687]
创新
[0688]
目前关于pd中alp基因的遗传变异的知识主要是对gba基因的研究。除了pd病例(n≈2700)和对照(n≈2000)的内部wes和外显子组芯片数据之外,这里描述的研究在来自外显子组聚合联盟(exac)(n≈61,000)的wes数据的大型公开可用的数据库中系统和全面地评估了alp的各基因中的遗传变异对发展pd风险的贡献。使用这些多重数据集,设计了一组分析,该组分析将揭示与pd相关联的alp功能障碍的遗传结构。
[0689]
关于特异性alp基因在pd发病机制中的作用,尚无共识。这部分地是由于以下事实:大多数遗传学研究集中于少数基因(例如,gba、smpd1和npc1),而基于细胞的研究采用经典的表达系统和药理学方法。此外,许多此类研究未评估适当的pd途径(α
‑
syn聚集相对于线粒体自噬)。这里描述的研究并入了整合框架,该整合框架将计算方法和实验数据相结合,以在体外验证选定alp基因的功能效应。基于细胞的测定由生化数据、活细胞测定、人pd病例和对照中的全基因组基因表达数据、来自不同阶段人pd病例的全基因组基因表达数据以及来自pd小鼠模型的全基因组基因表达数据补充。为了实现本研究的目标,已组建了协作团队,该团队在lsd和pd方面均有经过验证的生产力记录。
[0690]
实验方法
[0691]
在pd中富含罕见功能变体的alp基因
[0692]
通过挖掘公共数据库(参见生物信息学分析)和文献中的现有注释,手动编制了自噬
‑
溶酶体基因集(约430个基因)的列表。alp功能障碍的最佳研究模型是lsd。lsd是由纯合lof、复合杂合突变或拷贝数变异(cnv)引起的一组遗传异质性孟德尔疾病。使用了exac数据库(其含有>60,000个测序的外显子),以在广泛的族裔抽样中估计引起lsd的变体的频率。引起lsd的基因表现出广泛的蛋白改变变体,从npc2基因中的三十二种到gaa基因中的423种。这些变体中有许多被预测为lof,可能在杂合状态下表现为亚形变体。大多数pd样品来自欧洲血统。因此,这里呈现的分析集中在非芬兰样品(nfe,约33,000名个体)中的引起lsd的基因(n=46)。存在在ncbi clinvar数据库中报告的约2740种引起lsd的变体。存在在
exac样品中注释的288种引起lsd的变体:76%为错义变体,10%影响替代剪接,并且12%为无义突变。大部分引起lsd的变体通过sift被预测为有害的(87%),并且通过polyphen2被预测为危险的(84%)。大多数(73%)引起lsd的变体位于高度保守的核苷酸中(grep评分>4)。每个基因的这些变体(杂合携带者的数量)的积累次要等位基因频率(cmaf)的范围从cstd基因中的1.50e
‑
05
到npc2基因中的0.003。因此,应用1
×
10
‑3的cmaf阈值作为保守上界,因为在一般群体中比这更频繁出现的变体预计不会是高度外显变体。
[0693]
发现样品由来自ppmi组群的331例不相关pd病例的wes数据组成。表9显示了与pd相关联的顶级引起lsd的基因。
[0694]
表9.与nfe(exac)相比,在ppmi pd中的引起lsd的基因中发现的罕见变体
[0695]
基因cmaf病例maf exacor95%cip值glb10.00130.000373.52.8
‑
4.81.1e
‑
33ppt10.000910.000692.31.9
‑
3.84.8e
‑
08neu10.001510.0011.51.4
‑
6.91.8e
‑
06gba0.004320.002032.11.3
‑
3.36.19e
‑
06gns0.000760.000312.41.3
‑
21.52.57e
‑
05naglu0.00910.000194.71.5
‑
14.91.05e
‑
04
[0696]
根据exac样品中定义引起lsd的变体的特征,使用纳入和排除标准对它们进行分析。如所预期的,来自nfe exac数据集的基因特异性cmaf与来自内部pd数据库的cmaf高度一致(ppmi r2=0.92;wustl r2=0.96)。每个引起lsd的基因(n=46)中符合纳入标准的变体按基因进行核对:82%为错义变体,15%影响替代剪接,并且3%为无义突变。将罕见蛋白改变变体(cmaf)的负荷与在对照和exac中观察到的负荷进行比较。对于这些基因中的大多数,与对照相比,在病例中存在过多的变异,但未发现与内部对照之间的关联。当与exac样品的cmaf相比时,七个基因通过了严格的多重检验校正阈值p<1.0
×
10
‑3(0.05/50),并且三个引起lsd的基因通过了基因水平显著性阈值p<2.4
×
10
‑6(0.05/20,000)(参见,例如表9)。接下来,使用另一个pd组群(wustl)来重复表9中所列的发现,包括490例pd病例,其中使用人类外显子组芯片来获得数据(参见,例如表10)。
[0697]
表10.与exac(非芬兰裔欧洲人)相比,在wustl pd中的引起lsd的基因中发现的罕见变体
[0698][0699]
如所预期的,使用外显子组芯片数据在样品中复制了七个基因(参见,例如表10)。值得注意的是,在复制样品中发现的关联在相同的方向并且效应大小是相同的。为了支持这些遗传发现,在具有ppt1突变的患者中已经报告了黑质(sn)的pd和色素脱失。在伴有
naglu基因突变的mps iiib患者中已经报告了sn病理和路易体积累。此外,与对照相比,在来自pd患者的黑质的da神经元中发现naglu和ppt1基因的转录水平的降低(参见,例如图2,图20)(geo数据库;系列gds2821)。因此,基于先前发布的关于变体致病性的数据(clinvar数据库)、在一般群体(exac)和pd病例中的频率以及对蛋白影响的计算机预测,选择了naglu和ppt1基因的每一种中的三种变体以进行功能分析(参见,例如表11)。
[0700]
表11.用于功能验证的引起lsd的基因中的选定的罕见变体。
[0701][0702]
αsyn pff的制备和使用之前已在神经元培养中被优化。在体外(div)第7天,将αsyn pff添加到来自野生型小鼠的原代皮质神经元中。治疗后7天,固定神经元并用p
‑
α
‑
syn特异性抗体染色。pff诱导内源性表达的α
‑
syn募集为异常的、磷酸化的、不溶性聚集体(参见,例如图3a和图3b)。α
‑
syn聚集体最初在突触前终末和轴突中表现为小的点状内含物(参见,例如右下板图,图3a)。聚集体生长,并且外观变得更细长和蜿蜒,类似于路易神经突(参见,例如左下板图,图3a)。图3b显示,pbs处理的对照神经元显示出略高于15kda的条带,其对应于单体α
‑
syn。在用pff处理的神经元中出现了几条具有较高分子量的条带。那些额外的条带可能对应于α
‑
syn低聚物。这是研究溶酶体功能障碍对α
‑
syn聚集的影响的易处理体外系统。
[0703]
研究设计与方法
[0704]
(i)鉴定pd中富含罕见功能变体的alp基因
[0705]
假设,alp的基因中未经测试的罕见功能变体会影响发展pd的风险。这里,描述了整合方法,该方法将大数据集(内部和公共可用的数据库)中对alp基因的预测的罕见功能变体的分析相结合,以优先考虑alp中有证据表明参与ad风险的候选基因。(i)的方法和实验设计描述如下。
[0706]
样品和数据采集
[0707]
如这里所示出的,已经在pd中分析了引起lsd的基因。以下数据集也用于确定380个额外alp基因在pd中的作用。
[0708]
帕金森病进展标记物倡议(ppmi):ppmi数据集的遗传、表型和流行病学数据可在ppmi网站上获得。已获得所有ppmi样品的gwas、外显子组芯片和wes数据(参见,例如表12)。
[0709]
表12.pd可用的样品。
[0710]
研究对照病例mdc20001000ppmi200400un550654总计27502054
[0711]
此数据集以前曾用于多项研究。csf aβ、tau和α
‑
突触核蛋白水平对于所有参与者可获得。
[0712]
华盛顿大学运动障碍诊所(mdc)(movement disorder clinic(mdc)at washington university):可以从mdc获取样品,其包括来自超过3000个样品的临床、遗传和dna数据。已经获得了这些样品的gwas和外显子组芯片数据(但未获得wes数据)(参见,例如表12)。csf aβ、tau和α
‑
突触核蛋白水平目前对于300名参与者可获得。额外的pd病例和对照可以通过mdc诊所招募。mdc诊所每周平均评估10名新诊断的pd患者。估计每年可以招募约500名新的pd病例和对照。获得了所有新招募个体的dna、gwas和外显子组芯片数据。
[0713]
纳瓦拉大学(un)运动障碍研究中心(movement disorders unit at the university of navarra(un)):可以获取总计654例pd病例和550例pd对照的临床数据和遗传资料,其然后用于复制这里介绍的美国欧洲人组群中的发现。为所有这些样品生成了gwas和外显子组芯片数据。基于所选参与者的基因型为所选参与者生成wes/wgs。pd临床诊断基于英国脑库标准(uk brain bank criteria)。在登记之前,已获得所有参与者的书面知情同意。这三个群体的人口统计学特征已在之前发布。
[0714]
定义罕见变异的等位基因频率阈值
[0715]
使用从引起lsd的变体的分析中获得的信息(包括引起lsd的变体的最高maf、sift、polyphen2或gerp评分),定义了以下纳入标准:1)ad病例中的调用率>98%,2)每个变体的maf<0.01%,3)由exac或系综注释为错义的指定规范转录物中的仅可能的蛋白改变变体,4)框架移位,5)无义,6)影响剪接供体和受体区域的变体,和7)如果在ncbi clinvar数据库中有致病性证据并且位于3'或5'utr区域。在exac中未发现的变体,或ncbi clinvar数据库中不存在的同义、内含子、3'或5'utr变体或maf>0.01%、exac和clinvar中的错误调用被排除在外。为了确保群体特异性变体对本分析没有混杂效应,基于主组分结果选择了个体(参见群体结构分析)。
[0716]
数据下载自exac(版本0.3.1,2016年6月)
[0717]
在分析中仅包括具有覆盖至>30
×
的中位序列深度的高比例的编码区以及仅高质量(带通滤光器)变体的基因。
[0718]
gwas、qc和估算
[0719]
使用illumina 610、omniexpress芯片或人类核心外显子组对wustl和ppmi样品进行了基因分型。对基因型数据应用了严格的qc。如果(1)每个snp或每个个体的基因分型成功率<98%,(2)哈迪
‑
温伯格平衡(hwe)(p<1
×
10
‑
6)或(3)maf或缺失<0.05,则snp被剔除。qc基于基因分型芯片分别进行。在去除低质量snp和个体后,使用shapeit
‑
impute2管道利用1,000个基因组项目(第3阶段发布)的参考单倍型进行基因型估算。基因型估算是基于所用的基因型平台分别进行的。由impute2报告的或来自hwe的信息评分质量小于0.3、后验概率<0.9的snp被移除。使用illumina neurox2阵列对额外的样品进行基因分型。这是定制的
illumina人类核心外显子组阵列。illumina人类核心外显子组含有约226,000个选择用于估算的常见snp以及另外225,000个编码标记物。neurox2阵列还含有额外的自定义内容,包括所有已知的已报告的pd致病变体,以及来自gwas的pd的p值<10
‑3的所有信号的标记的snp。额外基因分型的样品的qc和估算遵循相同的管道。此外,可以使用hrc管道重新估算所有的样品。
[0720]
外显子组芯片qc
[0721]
使用之前描述的用于调用illumina外显子组芯片数据的最佳实践进行基因分型调用。外显子组芯片的qc与用于gwas的qc步骤相似,但由于低的maf,未去除变体。由于已获得外显子组芯片的原始数据,因此可以在单个变体或基因水平上检查聚类的任何显著关联。
[0722]
全外显子组测序数据
[0723]
已经用于qc和调用变体的方法可以用于这里所述的研究。简而言之,外显子组文库是使用安捷伦的sureselect制备的。序列数据在hiseq4000上生成,具有150
×
2对末端读数,其中平均覆盖深度为至少30
×
。针对grch37.p13基因组参考进行比对,尽管根据该基因组版本对gatk的支持,可能会将其更改为grch38。变体调用按照gatk 3.6的最佳实践执行。按照gatk的建议,对wes序列进行比对,并分别对变体调用。仅那些属于上述99.9部分的变量和插入缺失被考虑用于分析;确定了等位基因平衡(ab=0.3
‑
0.7)、质量(qual≥30)、读取深度(dp≥10)和缺失(geno=0.02)的变体阈值。从分析中移除超出哈迪
‑
温伯格平衡(p<1
×
10
‑
6)或病例与对照之间缺失差异的变体。此外,从数据集中移除缺失变体超过2%且来自临床数据库的基因型数据表明存在性别不一致的个体。最后,使用plink1.9和这些个体的现有gwas数据集确认个体和家族相关性。变体的功能作用和群体频率用snpeff标注。
[0724]
群体分层
[0725]
主组分分析是使用eigenstrat和hapmap样品作为锚定物进行的,以确认自我报告的种族/民族。仅包括欧洲裔的个体,以避免由于群体分层导致的虚假关联。
[0726]
注释
[0727]
使用seattleseq、外显子组变体服务器(exome variant server)、snp功能注释门户(snp function annotation portal)和pharmgkb进行snp注释。
[0728]
负荷测试
[0729]
使用之前开发的经验证的统计方法来分析与罕见变体的关联。简而言之,基于基因的方法将区域内的罕见变体折叠成单个值,并且然后测试区域内的罕见变体与感兴趣性状之间的关联。序列核关联检验(skat)用于检验基因区域内的状态与罕见变体之间的关联。该方法已经被成功地用于先前的研究中。
[0730]
这里呈现的结果和功效计算表明,在基于基因的分析中,有足够的功效来检测>2.7的平均优势比。如果病例对照设计未复制alp基因的关联,则可以使用内表型设计。因此,可以测试alp基因中的常见或罕见变体是否影响α
‑
突触核蛋白的脑脊液(csf)水平(800名个体)。基因组聚集数据库(126k外显子组)用于补充一般群体中alp基因的筛选。
[0731]
(ii)体外确定α
‑
突触核蛋白聚集体和线粒体自噬中特异性alp基因的功能效应假设多个alp基因中的特定缺陷有助于pd的发病机制。
[0732]
(ii)(a)的方法和实验设计描述如下。使用该方法,可以确定naglu和ppt1基因中
选定的变体对酶活性、蛋白和rna水平以及溶酶体功能的影响。
[0733]
(ii)(a)概述
[0734]
评估(i)中鉴定的与pd相关联的每种alp基因的所有变体超出了这里所述研究的范围。因此,重点关注在aim 1中鉴定的3种变体(参见,例如表11)。已经使用严格的标准来选择naglu和ppt1基因(之前被确定为致病变体的两种)中的潜在功能变体。然而,表征它们对mrna稳定性、蛋白水平和酶活性的影响非常重要。为此,生成了来自naglu和ppt1敲除小鼠的原代细胞,并用表11中的变体进行转导,并且确定了它们对酶活性、蛋白和rna水平的影响。还确定了对溶酶体功能的影响。naglu和ppt1基因的cdna已经在质粒中获得,以用于在细菌和哺乳动物细胞中表达。使用定点诱变试剂盒(quikchange ii(安捷伦科技,美国加利福尼亚州圣克拉拉)对选定的变体进行工程改造。
[0735]
慢病毒产生
[0736]
野生型和突变型cdna被亚克隆到plenti
‑
iii
‑
pgk载体(应用生物材料公司,加拿大里士满)中,该载体携带嘌呤霉素抗性基因。如前所述,将所得的慢病毒载体与编码vsv
‑
g、gag
‑
pol和rev的质粒一起共转染到hek
‑
293t包装细胞中。根据先前公布的方案收集病毒上清液。将细胞暴露于未浓缩的病毒上清液持续24小时,并用5μg/ml的嘌呤霉素选择细胞持续四周。如前所述进行原代神经元培养。
[0737]
mrna和蛋白水平的影响
[0738]
使用特异性引物进行定量实时pcr,以测试所选的变体对mrna水平和剪接的影响。使用以下抗体进行蛋白免疫印迹:抗ppt1(genetex)和抗naglu(ab137685,abcam)。
[0739]
基于细胞裂解物的溶酶体酶活性
[0740]
为了确定变体对酶活性的影响,如前所述,进行了naglu和ppt1活性的荧光测定。简而言之,在日立f
‑
2000荧光分光光度计(日立,加利福尼亚州普莱森顿)中,使用范围为0.02至5mm的4
‑
甲基伞形酮(sigma公司,密苏里州圣路易斯)的标准曲线,在448nm发射和365nm激发下测量4
‑
甲基伞形酮基
‑
n
‑
乙酰基
‑
α
‑
氨基葡萄糖苷(naglu)和4
‑
甲基伞形酮基
‑6‑
硫代棕榈酰基
‑
β
‑
d
‑
葡萄糖苷(ppt1)裂解。
[0741]
溶酶体功能
[0742]
通过使用溶酶体跟踪器使用流式细胞术定量每个细胞的酸性隔室的数目,来确定变体是否正影响全局溶酶体功能。溶酶体膜完整性通过1mm的llome,然后通过galactein
‑
3染色进行监测(sc
‑
23938)。
[0743]
(ii)(b)的方法和实验设计描述如下。使用这种方法,可以确定naglu和ppt1基因对α
‑
syn聚集的影响。
[0744]
(ii)(b)概述
[0745]
可以确定来自naglu和ppt1缺陷及半合子小鼠的原代皮质和海马神经元中内源性α
‑
syn的清除率是否存在变化。慢病毒载体用于在来自naglu和ppt1缺陷小鼠的原代神经元中过表达α
‑
syn,并测试其对清除率和神经毒性的影响。然后,在用外源性α
‑
syn预制原纤维(pff)处理后,从naglu和ppt1缺陷小鼠中测定原代神经元中α
‑
syn聚集体(内含物)的速率和水平(参见,例如图3a和图3b)。最后,确定重组酶替代是否可以减轻来自naglu和ppt1缺陷小鼠的神经元中α
‑
syn预制原纤维的毒性。
[0746]
α
‑
突触核蛋白(αsyn)预制原纤维(pff)的生成
[0747]
生成pff的条件已在之前被优化。小鼠和人野生型αsyn单体蛋白如前所述在大肠杆菌中产生。简而言之,用克隆到prk172细菌表达载体中的αsyn转化bl21 de3 ril感受态大肠杆菌。将细菌颗粒重新悬浮在高盐缓冲液中,使用kontes均质机进行均质化,声处理,并且煮沸以沉淀不需要的蛋白。离心所得的裂解物,并透析上清液,并且通过尺寸排阻和阴离子交换色谱法纯化。为了生成αsyn pff,将单体蛋白以5mg/ml的浓度在37℃下在以1000rpm振荡的热混合器(thermomixer)中在tris缓冲的nacl中孵育72小时。为了测定最终pff的浓度,将一小等份所得的pff混悬液以17,000x g离心15分钟,并通过bca测定来确定上清液的总蛋白浓度,并且从振荡前取出的等分试样的浓度中减去。pff在tris缓冲的nacl中稀释,并在5
‑
10div后加入到培养的原代神经元中。暴露4
‑
7天后,通过免疫荧光或顺序提取和免疫印迹来确认pff转导和接种。使用抗
‑
磷酸
‑
α
‑
突触核蛋白(ser129)抗体,克隆81a(biolegend,mms
‑
5091),经由免疫荧光来检测来自内源性α
‑
syn的异常α
‑
syn聚集体。pff处理的神经元用突触前(cspα或snap
‑
25)、内吞(eea1)、自噬体(rab7)和溶酶体(lamp1)标记物共同染色。确定在溶酶体中是否存在αsyn聚集体的富集。使用组织和培养细胞溶酶体富集试剂盒(lysosome enrichment kit for tissue and cultured cells)(赛默飞世尔科技公司)进行亚细胞分级,使用lamp1、rab7和eea1作为溶酶体、晚期和早期内体标记物的对照。
[0748]
巨噬细胞的药理学调节
[0749]
如这里所述,可以确定自噬途径的药理学调节是否可改善α
‑
syn清除率。用自噬系统的激活剂和抑制剂(torin1,3
‑
甲胺、布雷非德菌素a和spautin
‑
1)以及伴侣介导的自噬(ar7)处理转导的和pff处理的细胞。使用lc3和p62的蛋白免疫印迹作为巨噬细胞活化的间接指标。使用溶酶体营养剂诸如氯喹、氯化铵(nh4cl)和亮抑蛋白酶肽作为阳性对照。
[0750]
重组酶
[0751]
已经获得足够的重组naglu和ppt1,用于体外添加到缺陷神经元中。在存在或不存在摄取/结合抑制剂(甘露糖
‑6‑
磷酸:m3655,sigma)的情况下,在暴露于αsyn pff之前、期间和之后添加酶。
[0752]
(ii)(c)的方法和实验设计描述如下。如这里所述,该方法可以确定naglu和ppt1基因对线粒体自噬的影响。
[0753]
(ii)(c)概述
[0754]
如这里所述,可以确定在来自naglu和ppt1缺陷小鼠的原代多巴胺能神经元中是否存在线粒体自噬的变化。如前所述进行原代多巴胺能神经元培养。
[0755]
线粒体自噬测定
[0756]
在存在或不存在20μm羰基氰化物m
‑
氯苯腙(cccp)(sigma,c2759)和/或巴弗洛霉素a1(sigma,b1793)的情况下孵育细胞12
‑
24小时。线粒体跟踪器(mitotracker)和jc
‑
1染料以及与lc
‑
3、sqstm1和lamp
‑
1蛋白的共定位用于评估线粒体自噬。通过蛋白免疫印迹来检测线粒体蛋白(tom20,细胞色素c,复合物iii(c
‑
iii)核心1,tim44)降解。在来自naglu和ppt1缺陷小鼠的原代多巴胺能神经元中,通过qpcr和蛋白免疫印迹来检查parkin和pink1水平。
[0757]
可替代地,基因组编辑方法(诸如crispr技术)可以用于在naglu和ppt1基因中引入所选的变体,并进行上述功能测定。在存在α
‑
syn pff的情况下,通过过表达tfeb可以增
加总溶酶体活性。电子显微镜可以用于测量对线粒体自噬的影响。利用本项目生成的数据将成为被设计成确定naglu和ppt1基因在体内pd发病机制中的作用的更彻底实验的基础。
[0758]
实例6:阿尔茨海默病(ad)和帕金森病(pd)的新治疗性分子靶的鉴定
[0759]
本实例描述了用于治疗阿尔茨海默病(ad)和帕金森病(pd)的新治疗靶的鉴定。
[0760]
如本文所公开的,鉴定了在患有阿尔茨海默病的患者的样品中的至少12个溶酶体基因中具有较大的效应大小(or>2.5)的预测功能变体的富集。
[0761]
表13.与对照相比,在ad中引起lsd的基因中发现的罕见变体的总结。
[0762]
[0763][0764]
表14.与对照相比,pd中引起lsd的基因中发现的罕见变体的总结。
[0765][0766]
大约十分之一的人在其一生的某个时候会遭受痴呆。目前,美国有超过800万人患有某种形式的痴呆。随着人口老龄化,这一数字预计将大幅增加。百分之六十到八十的痴呆患者(约500万)将被临床诊断为阿尔茨海默病(ad)。然而,只有一半诊断为ad的患者实际上将具有ad样病理。因此,约250万美国人患有不属于任何已知分类的痴呆形式。帕金森病(pd)是仅次于ad的第二常见神经退行性疾病,目前美国有多达一百万人患有pd。
[0767]
越来越多的证据表明,严重的溶酶体功能障碍可能在几种神经退行性疾病(最显著的是ad和pd)中发挥作用。目前认为,溶酶体基因完全或几乎完全功能丧失的变体将与病因不明的痴呆病例相关联。此时,已有少数单个溶酶体基因与分离群体中的ad或pd相关联。直接比较了ad患者(n=1,394)和pd患者(n=821)与欧洲裔对照个体(n>33,000)的代表性样品之间的溶酶体基因有害变体的频率。使用非常严格的统计方法,鉴定出20个和8个溶酶体基因中的罕见功能变体,这些变体分别在ad和pd病例中显著过量表达(参见,例如表13和表14)。考虑到仅在遗传分析中鉴定的罕见变体的计算的携带者频率,估计540万ad患者中至少有3%和100万pd患者中至少有2%可能具有那些基因的缺陷。具有溶酶体基因缺陷的ad和pd患者的实际数量甚至可能更多。
[0768]
溶酶体贮积病(lsd)是一类相对较大的遗传性代谢疾病,其涵盖至少50种不同的疾病。这些疾病由单个溶酶体基因中的完全或几乎完全的功能丧失突变引起,并且通常以常染色体隐性方式遗传。针对lsd的有效疗法取得了重大的进展。治疗途径包括小分子伴侣、终止密码子通读药物、底物减少、基因疗法、酶替代、干细胞介导的疗法等。尽管lsd通常
被认为是儿科神经退行性疾病,但数据表明溶酶体基因缺陷与成人发作的神经退行性疾病的病例有关。
[0769]
这里呈现的数据表明,在相当大比例的痴呆病例中,溶酶体基因缺陷与lsd疗法的发展相结合,该数据具有以下含义:
[0770]
1)遗传数据可以构成患有ad或pd的患者的诊断筛查工具的基础。
[0771]
2)具有与ad和pd相关联的溶酶体基因的变体的患者可能潜在地用上述所列的疗法之一治疗,以停止或逆转疾病。
[0772]
3)在溶酶体基因中携带神经变性相关变体的症状前家族成员可以在疾病发作之前或在疾病的最早阶段进行治疗。
[0773]
4)对于成人发作的神经退行性障碍,重新使用lsd有效疗法可以被视为疾病缓解。
[0774]
实例7:与成人发作的神经系统疾病相关联的溶酶体/自噬基因变体
[0775]
本实例描述了与成人发作的神经系统疾病相关联的溶酶体/自噬基因中的遗传变体,以及在体内测试这些变体的效果的实验和策略。
[0776]
背景
[0777]
年龄是阿尔茨海默病(ad)和帕金森病(pd)的发展和进展的最大危险因素。衰老也会降低自噬
‑
溶酶体途径(alp)的降解能力。尽管多项体外和体内研究表明alp功能障碍有助于ad和pd的发病机制,但尚不完全了解alp功能的年龄依赖性和ad相关联下降背后的遗传变异。
[0778]
假设
[0779]
主要假设是,在溶酶体/自噬基因中携带杂合遗传变体的个体有发展常见成人发作的神经系统疾病(例如,阿尔茨海默病、帕金森病、额颞叶痴呆等)的风险。此外,通过酶替代疗法(ert)、基因疗法(gt)、干细胞疗法等补充外源性溶酶体蛋白将减缓这些疾病的进程。
[0780]
遗传关联
[0781]
阿尔茨海默病
[0782]
在4,000例ad病例和5,000名内部对照中,对所有alp基因(n=430)进行了单个变体和基于基因的分析。在来自阿尔茨海默病测序项目(adsp)的独立样品(5,000例ad病例和4,500名对照)中复制了这些结果。使用来自外显子组聚合联盟(exac)数据库的非芬兰欧洲人(n=33,350)的wes数据分析了所有alp基因,以确定来自欧洲血统个体的alp的每个基因中的基线遗传变异。
[0783]
帕金森病
[0784]
在1,000例pd病例和1,000名内部对照中,对所有alp基因(n=430)进行了单个变体和基于基因的分析。在来自国际帕金森病遗传学联合会(ipgdc)的独立样品(5,500例pd病例和6,000名对照)中复制了这些结果。
[0785]
结果
[0786]
对于大多数基因,与内部对照相比,在病例中存在过多的变异,但仅发现与几个基因之间的名义关联。当将ad或pd病例的cmaf与exac样品进行比较时,几个独立基因通过了多重检验校正阈值。接下来,使用adsp或ipgdc组群来重复这些发现。在这两个独立样品中复制了几个基因。
[0787]
支持数据
[0788]
a)mrna水平
[0789]
1.人类
[0790]
2.小鼠模型
[0791]
b)间质液
[0792]
体内概念验证
[0793]
a)阿尔茨海默病:
[0794]
1.naglu+/
‑
;5xfad
[0795]
2.ppt1+/
‑
;5xfad
[0796]
3.dnajc5+/
‑
;5xfad
[0797]
b)帕金森病:
[0798]
1.注射有α
‑
突触核蛋白的预制原纤维(pff
‑
αsyn)的naglu
‑
/
‑
[0799]
2.注射有pff
‑
αsyn的ppt1
‑
/
‑
[0800]
3.注射有pff
‑
αsyn的galc
‑
/
‑
和+/
‑
[0801]
4.注射有pff
‑
αsyn的acd
‑
/
‑
和+/
‑
[0802]
挽救实验
[0803]
基因疗法可以挽救由于溶酶体酶基因中的杂合突变引起的溶酶体功能障碍。单次干预将在cns中产生持续、广泛的影响。
[0804]
a)阿尔茨海默病:
[0805]
1.naglu+/
‑
;5xfad
[0806]
a)加上gt
[0807]
2.ppt1+/
‑
;5xfad
[0808]
a)加上ert
[0809]
b)加上gt
[0810]
3.dnajc5+/
‑
;5xfad
[0811]
a)加上gt
[0812]
b)帕金森病:
[0813]
1.注射有α
‑
突触核蛋白的预制原纤维(pff
‑
αsyn)的naglu
‑
/
‑
[0814]
a)加上gt
[0815]
2.注射有pff
‑
αsyn的ppt1
‑
/
‑
[0816]
a)加上ert
[0817]
b)加上gt
[0818]
3.注射有pff
‑
αsyn的galc
‑
/
‑
[0819]
a)加上ert
[0820]
b)加上gt
[0821]
淀粉样蛋白积累结果
[0822]
本文表明,β
‑
淀粉样蛋白积累在对于ppt和naglu半合的小鼠中加剧(参见,例如图14a、图14b)。
[0823]
半合子幼稚naglu小鼠在脑间质液(isf)中显示出较低的aβ水平。同窝wt和半合子
小鼠中aβ的isf水平的微透析定量显示aβ的基线isf水平的显著降低。半合子幼稚ppt1小鼠在脑间质液(isf)中表现出较低的aβ水平。同窝wt和半合子小鼠中aβ的isf水平的微透析定量显示aβ的基线isf水平的显著降低。
[0824]
aβ斑块负荷结果
[0825]
表明,ad(5xfad+/
‑
)模型中的naglu、ppt1和dnajc5半合子性会加剧β淀粉样蛋白的积累。(a)5xfad+/
‑
、(b)5xfad+/
‑
/naglu+/
‑
、(c)5xfad+/
‑
/ppt+/
‑
和(d)5xfad+/
‑
/dnajc5+/
‑
海马区在7个月时的β
‑
淀粉样蛋白染色的代表性图片(参见,例如图21a至图21d)。
[0826]
还表明ppt1和naglu基因的半合子性加剧了β
‑
淀粉样蛋白的积累(参见,例如图21e)。与5xfad小鼠相比,在ppt1半合子/5xfad小鼠中由斑块覆盖的表面积增加。海马中斑块负荷的量化是盲法进行的。通过不成对t检验,**p<0.01。7个月大的5xfad(n=4)和ppt1半合子/5xfad(n=8)。与5xfad小鼠相比,在naglu半合子/5xfad小鼠中由斑块覆盖的表面积增加。海马中斑块负荷的量化是盲法进行的。通过不成对t检验,*p<0.05。7个月大的5xfad(n=4)和naglu半合子/5xfad(n=4)。
[0827]
生存结果
[0828]
本文已经证明,靶向的aav2/9
‑
ppt1基因疗法延缓ppt1半合子/5xfad小鼠中的疾病进展。kaplan
‑
meier生存曲线显示,未经治疗的ppt1半合子/5xfad(红色,n=8)的中位生存期明显短于颅内用aav9
‑
ppt1治疗的半合子/5xfad(绿色,n=5)。采用对数秩检验的趋势分析对总生存期有显著意义(p<0.0001)。按照所有说法,用aav2/9
‑
ppt1治疗的小鼠都是健康的,但在第7.8个月时特意将其处死以进行组织学分析。
[0829]
实例6:lsd基因的携带者家族的病例研究
[0830]
本实例描述了对具有lsd(婴儿神经元性蜡样脂褐质病,cln1疾病)的儿童的家族的研究。cln1疾病由溶酶体酶ppt1(棕榈酰蛋白硫酯酶1)的纯合子缺失引起。根据定义,双亲对ppt1基因中的完全或几乎完全功能丧失突变是杂合的(携带者)。因此,我们获得了创建母亲和父亲家庭谱系(参见,例如图24)的书面同意。家族史显示ad及其他相关形式的成人神经系统疾病和/或痴呆的增加。特别注意的是,ad的正式诊断只能在尸检后做出。在临床上,ad可能呈现出类似于其他形式的成人发作的神经系统疾病的表现。该家系中的许多受影响的家庭成员尚未获得正式诊断(即未进行尸检)。然而,ppt1基因中的杂合性完全或几乎完全功能丧失突变已经通过该家族传了几代。该家族中的神经系统疾病和/或痴呆的显著增加与上述遗传和生物学数据完全一致。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1