一种复方利多卡因乳膏中丙胺卡因氧化杂质的分离纯化方法与流程
本发明涉及药物分析和药物制剂领域,具体涉及一种复方利多卡因乳膏中丙胺卡因氧化杂质的分离纯化方法。
背景技术:
在新药及仿制药研究中,国内外对杂质的研究越来越被重视,ich和nmpa都相继发布了杂质研究的指导原则,国外药典以及中国药典都对杂质作了专门的规定,而药品研究中所涉及的杂质包括原料药以及制剂中所包含的。对于企业而言,新药和仿制药的报批过程中杂质研究也越来越受重视,对于杂质的研究越透彻,被审评通过的几率就会大大增加,因此杂质的研究对于药品研发过程中所占的比重越来越大,而杂质的分离制备是药物研究中最为关键的技术之一。
丙胺卡因(prilocaine),化学名n-(2-甲基苯基)-2-丙胺-丙酰胺,分子式为c13h20n2o,cas号为721-50-6。丙胺卡因是一种用于硬膜外、阻滞和浸润等各种手术麻醉的药物,而现在市场上多采取复方制剂,其中包含利多卡因和丙胺卡因两种活性成分。在对复方利多卡因制剂的质量研究中发现一个超标的未知杂质,影响了产品质量,需要进行质量控制。
技术实现要素:
为解决目前复方利多卡因质量控制中存在的问题,本发明提供一种复方利多卡因乳膏中丙胺卡因氧化杂质的分离纯化方法,本发明通过运用制备液相色谱、液相色谱质谱联用(lc-ms)、核磁共振谱(nmr)、冷冻干燥机等多种技术,对此种杂质进行了分离、制备、纯化、冻干、检测和鉴定,此种杂质可以在复方利多卡因乳膏制剂质量检测中作为杂质对照品。
本发明的技术方案如下:
一种复方利多卡因乳膏中丙胺卡因氧化杂质的分离纯化方法,其特征是分离出的杂质分子式为c15h22n2o,结构式如下:
一种复方利多卡因乳膏中丙胺卡因氧化杂质的分离纯化方法,其特征是包括以下步骤:步骤(1)破坏实验:称取丙胺卡因原料药溶于水中,加入氧化铁粉末,140℃下搅拌加热10小时,待体系冷却后,随后加入流动相溶液,充分混合,过滤待用;
步骤(2)制备液相色谱法分离制备杂质:通过连续三次分离纯化步骤,且每次分离纯化得到的溶液都通过冷冻干燥机干燥后再重新上样。在制备液相色谱的工作站中,收集所需要的此种杂质,并且全程通过液相色谱质谱联用(lc-ms)对分离纯化过程进行跟踪监测;
步骤(3)纯化:对步骤(2)中通过液相色谱质谱联用(lc-ms)监测收集到的分离组分,通过冷冻干燥机进行冻干,得到此种杂质,并且于低温下保存。
所述步骤(1)所述的丙胺卡因原料药与氧化铁粉末的质量比为1:0.5;步骤(1)所述的流动相溶液为甲醇和水,其中甲醇与水的体积比为9:1,丙胺卡因原料药与流动相的质量体积比为1:100;
所述步骤(2)所述的制备液相的条件为:
第一次分离纯化:
仪器:gilsongx-281制备液相色谱仪和waters2767制备液相色谱仪;
色谱柱:welchxtimatec18柱(50×250mm,10μm);
流动相:甲醇-水溶液,甲醇:水=70-85:30-15(v/v)梯度洗脱;
柱温:室温;
流速:60ml/min;
进样体积:5ml;
压力:29bar;
检测波长:232nm;
第二次分离纯化:
仪器:waters2767制备液相色谱仪;
色谱柱:welchxtimatec18柱(30×150mm,5μm);
流动相:乙腈-水溶液,乙腈:水=30-50:70-50(v/v)梯度洗脱;
柱温:室温;
流速:35ml/min;
进样体积:1ml;
压力:1480psi;
检测波长:232nm;
第三次分离:
仪器:waters2767制备液相色谱仪;
色谱柱:welchxtimatec18柱(30×150mm,5μm);
流动相:乙腈-水溶液,乙腈:水=30-65:70-35(v/v)梯度洗脱;
柱温:室温;
流速:35ml/min;
进样体积:0.5ml;
压力:1450psi;
检测波长:232nm。
所述步骤(3)中收集到的组分通过冷冻干燥机冻干得到,冻干条件为-86℃、0.02mpa;样品纯度鉴定条件为,仪器:waters液相色谱质谱联用仪;分析柱:innovalods-2,4.6×250mm,5μm;流动相:甲醇:水=70:30(v/v),其中水为含0.1%氨水的水溶液,等度洗脱;柱温:室温;流速:1ml/min;进样体积:50μl;检测波长:232nm;所制备杂质的纯度大于95%;
一种复方利多卡因乳膏中丙胺卡因氧化杂质的分离纯化方法,其特征是所述复方利多卡因乳膏中丙胺卡因氧化杂质的结构鉴定为:
所收集的杂质组分采用质谱(ms)以及核磁共振谱(nmr)对杂质进行分析,最后确证其分子式和结构式,分子式为c15h22n2o,结构式如下:
本发明的有益效果:
本发明通过制备液相色谱、液相色谱质谱联用仪(lc-ms)、质谱(ms)和核磁共振谱(nmr)、冷冻干燥等技术,对此种杂质进行了分离、纯化、冻干、检测和鉴定,对于复方利多卡因乳膏制剂的质量控制具有重要的意义。
附图说明
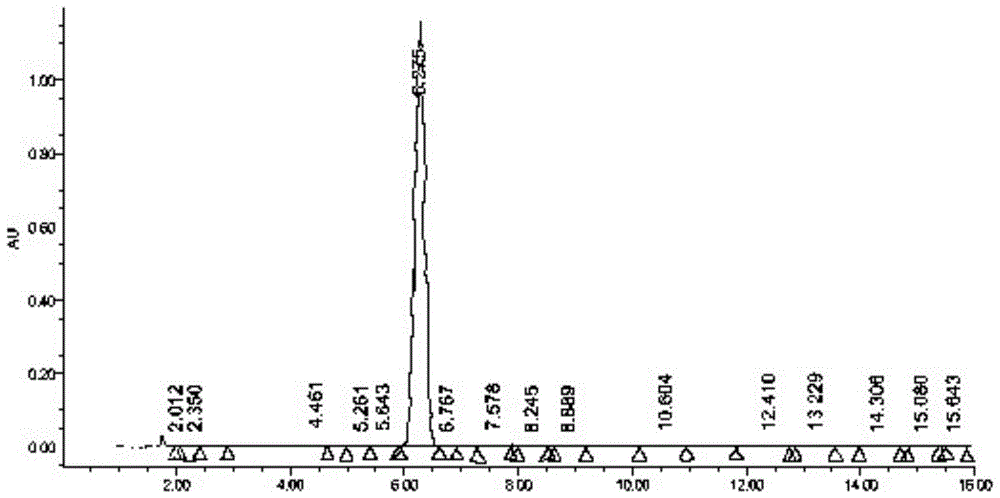
图1为本发明的丙胺卡因氧化破坏样品高效液相色谱图。
图2为本发明的第一次分离纯化制备液相色谱图。
图3为本发明的第二次分离纯化制备液相色谱图。
图4为本发明的第三次分离纯化制备液相色谱图。
图5为本发明的丙胺卡因氧化杂质纯度鉴定色谱图。
图6为本发明的丙胺卡因氧化杂质质谱图。
图7为本发明的丙胺卡因氧化杂质氢谱图。
图8为本发明的丙胺卡因氧化杂质h-hcosy图。
图9为本发明的丙胺卡因氧化杂质hsqc图。
图10为本发明的丙胺卡因氧化杂质hmbc图。
图11为本发明的丙胺卡因氧化杂质碳谱图。
图12为本发明的丙胺卡因氧化杂质碳谱dept90图。
图13为本发明的丙胺卡因氧化杂质碳谱dept135图。
具体实施方式
下面结合实施例对本发明作进一步的详细说明:
如图1至图13。
一种复方利多卡因乳膏中丙胺卡因氧化杂质的分离纯化方法,分离出的杂质分子式为c15h22n2o,结构式如下:
一种复方利多卡因乳膏中丙胺卡因氧化杂质的分离纯化方法,包括以下步骤:
步骤(1)破坏实验:称取丙胺卡因原料药溶于水中,加入氧化铁粉末,140℃下搅拌加热10小时,待体系冷却后,随后加入流动相溶液,充分混合,过滤待用;
步骤(2)制备液相色谱法分离制备杂质:通过连续三次分离纯化步骤,且每次分离纯化得到的溶液都通过冷冻干燥机干燥后再重新上样。在制备液相色谱的工作站中,收集所需要的此种杂质,并且全程通过液相色谱质谱联用(lc-ms)对分离纯化过程进行跟踪监测;
步骤(3)纯化:对步骤(2)中通过液相色谱质谱联用(lc-ms)监测收集到的分离组分,通过冷冻干燥机进行冻干,得到此种杂质,并且于低温下保存。
所述步骤(1)所述的丙胺卡因原料药与氧化铁粉末的质量比为1:0.5;步骤(1)所述的流动相溶液为甲醇和水,其中甲醇与水的体积比为9:1,丙胺卡因原料药与流动相的质量体积比为1:100;
所述步骤(2)所述的制备液相的条件为:
第一次分离纯化
仪器:gilsongx-281制备液相色谱仪和waters2767制备液相色谱仪;
色谱柱:welchxtimatec18柱(50×250mm,10μm);
流动相:甲醇-水溶液,甲醇:水=70-85:30-15(v/v)梯度洗脱;
柱温:室温;
流速:60ml/min;
进样体积:5ml;
压力:29bar;
检测波长:232nm;
第二次分离纯化
仪器:waters2767制备液相色谱仪;
色谱柱:welchxtimatec18柱(30×150mm,5μm);
流动相:乙腈-水溶液,乙腈:水=30-50:70-50(v/v)梯度洗脱;
柱温:室温;
流速:35ml/min;
进样体积:1ml;
压力:1480psi;
检测波长:232nm;
第三次分离
仪器:waters2767制备液相色谱仪;
色谱柱:welchxtimatec18柱(30×150mm,5μm);
流动相:乙腈-水溶液,乙腈:水=30-65:70-35(v/v)梯度洗脱;
柱温:室温;
流速:35ml/min;
进样体积:0.5ml;
压力:1450psi;
检测波长:232nm。
所述步骤(3)中收集到的组分通过冷冻干燥机冻干得到,冻干条件为-86℃、0.02mpa;样品纯度鉴定条件为,仪器:waters液相色谱质谱联用仪;分析柱:innovalods-2,4.6×250mm,5μm;流动相:甲醇:水(含0.1%氨水)=70:30(v/v),等度洗脱;柱温:室温;流速:1ml/min;进样体积:50μl;检测波长:232nm;所制备杂质的纯度大于95%;
一种复方利多卡因乳膏中丙胺卡因氧化杂质的分离纯化方法,其特征是所述复方利多卡因乳膏中丙胺卡因氧化杂质的结构鉴定为:
所收集的杂质组分采用质谱(ms)以及核磁共振谱(nmr)对杂质进行分析,最后确证其分子式和结构式,分子式为c15h22n2o,结构式如下:
制备液相色谱条件
我们通过对制备柱规格型号、流动相组合及配比、ph值、流速以及进样量进行了优化,得到以下分离条件。
第一次分离制备液相条件:welchxtimatec18,50×250mm,10μm,甲醇:水=70-85:30-15(v/v),梯度洗脱,进样体积:5ml,检测波长:232nm,流速:60ml/min;
第二次分离制备液相条件:welchxtimatec18,30×150mm,5μm,乙腈:水=30-50:70-50(v/v),梯度洗脱,进样体积:1ml,检测波长:232nm,流速:35ml/min;
第二次分离制备液相条件:welchxtimatec18,30×150mm,5μm,乙腈:水=30-65:70-35(v/v),梯度洗脱,进样体积:0.5ml,检测波长:232nm,流速:35ml/min;
制备监测:实验过程中全程通过lc-ms跟踪监测,确保分离纯化最终得到的杂质准确无误,采用waters液相色谱质谱联用仪为整个实验过程提供色谱法监测。每次制备液相进样后,收集到的杂质组分均通过lc-ms监测,确保组分收集准确、时间窗口合适、可以批量制备,杂质出峰良好。
监测液相条件为:innovalods-2,4.6×250mm,5μm,甲醇:水(含0.1%氨水)=70:30(v/v),等度洗脱,进样体积:50μl,检测波长:232nm,流速:1ml/min)
冷冻干燥:将通过lc-ms监测得到的合格组分,先进行预冻,随后在-86℃以及0.02mpa压力下的冷冻干燥机上进行冻干,产品低温保存。
本发明通过制备液相色谱、液相色谱质谱联用仪(lc-ms)、质谱(ms)和核磁共振谱(nmr)、冷冻干燥等技术,对此种杂质进行了分离、纯化、冻干、检测和鉴定,对于复方利多卡因乳膏制剂的质量控制具有重要的意义。
以上显示和描述了本发明的基本原理、主要特征和本发明的优点。本行业的技术人员应该了解,本发明不受上述实施例的限制,上述实施例和说明书中描述的仅为本发明的优选例,并不用来限制本发明,在不脱离本发明精神和范围的前提下,本发明还会有各种变化和改进,这些变化和改进都落入要求保护的本发明范围内。本发明要求保护范围由所附的权利要求书及其等效物界定。
- 还没有人留言评论。精彩留言会获得点赞!