一种UPLC-Q-TOF-MS快速筛查甘草中活性成分的方法与流程
一种uplc-q-tof-ms快速筛查甘草中活性成分的方法
技术领域
1.本发明属于医药分析领域,涉及一种uplc-q-tof-ms快速筛查甘草中活性成分的方法。
背景技术:
2.甘草为豆科植物甘草、胀果甘草或光果甘草的干燥根及根茎。外皮松紧不一,表面呈红棕色或灰棕色,气微,味甜而特殊。甘草具有清热解毒、祛痰止咳、补脾益气,缓急止痛,调和诸药等功效,其药效显著,在临床上使用频率非常高,且甘草在2019年底爆发的全球新型冠状病毒肺炎防治过程中起到了重要作用。我国甘草主要分布在宁夏、新疆、内蒙、甘肃等地,不同产地或品种的甘草中活性成分的种类及含量差异较大,因此准确定性识别甘草中的活性成分并定量检测,对于相关产品的产地溯源和质量分级是十分重要的。
3.目前,国内外研究多采用液相色谱、液相色谱串联质谱等方法对一种或多种甘草中活性成分进行定性定量分析,但鲜有利用uplc-q-tof-ms快速筛查甘草中活性成分的相关研究报道,且没有方法可实现20种以上甘草中活性成分的快速高通量筛查。但是,甘草及其制品的产地溯源、真假鉴别和质量分级都迫切需要其活性成分的高通量筛查检测技术。
4.因此亟待开发一种高通量的甘草中活性成分的快速筛查检测方法。
技术实现要素:
5.为解决上述问题,本发明提供了一种uplc-q-tof-ms快速筛查甘草中活性成分的方法。本发明以75%乙醇作为提取溶剂,其对甘草中各活性成分的提取率较高,稳定性较好,并且能有效去除干扰杂质,同时采用uplc-q-tof-ms对样品进行无标准品快速筛查。
6.本发明的具体技术方案为:一种uplc-q-tof-ms快速筛查甘草中活性成分的方法,包括以下步骤:
7.(1)样品前处理:将样品粉碎、过60目不锈钢筛网,称取约0.5g于离心管中,加入约50ml75%乙醇溶解,涡旋混匀;超声提取10min,然后再8000r/min离心5min,取上清液过0.22μm有机滤膜,收集滤液,待测;
8.(2)样品溶液经uplc分离,q-tof-ms采集数据,并基于各活性成分裂解规律,建立筛查谱库。
9.(3)uplc的分离条件为色谱柱acquity uplc hss t3,100mm
×
2.1mm,1.8μm;流动相a相为含0.1%甲酸水溶液,b相为乙腈,梯度洗脱;梯度洗脱的比例为0~10min,a为90%
→
70%;10~30min,a为70%
→
50%;30~40min,a为50%
→
10%;40~53min,a为10%;53~55min,a为10%
→
90%;55~60min,a为90%;流速0.5ml/min,进样量5μl,柱温45℃。
10.(4)q-tof-ms条件为esi离子源,负离子模式;离子源温度100℃;脱溶剂气温度550℃;脱溶剂气流速600l/h;碰撞能量:低碰撞能量off,高碰撞能量20~40v;毛细管电压2.0kv;锥孔电压40ev;锥孔气流速50l/h;扫描模式为灵敏度模式,扫描范围m/z 100~1200;扫描时间1.5s;数据采集模式为ms
e
模式。
11.自建筛查谱库包含甘草中20种活性成分的准分子离子、3个以上二级碎片信息以及保留时间等,其中阿魏酸准分子离子m/z为193.0501,二级碎片m/z有147.0452、163.0401、135.0452、145.0295、134.0373,保留时间为1.69min;异甘草苷芹糖准分子离子m/z为549.1608,二级碎片m/z有135.0088、119.0502、297.0768、255.0663、399.1085,保留时间为4.57min;甘草苷准分子离子m/z为417.1186,二级碎片m/z有255.0663、135.0088、119.0502、297.0768、399.1085,保留时间为4.57min;甘草苷元准分子离子m/z为549.1608,二级碎片m/z有255.0663、135.0088、297.0768、119.0502、417.1191,保留时间为4.59min;新甘草苷准分子离子m/z为417.1186,二级碎片m/z包含255.0663、135.0088、119.0502、150.0322、269.0456,保留时间为7.58min;异甘草素准分子离子m/z为255.0657,二级碎片m/z有119.0502、135.0088、149.0244、150.0322、117.0346,保留时间为8.07min;芒柄花素准分子离子m/z为267.0657,二级碎片m/z有252.0428、251.0350、132.0217、135.0088、119.0502,保留时间为9.33min;芒柄花苷准分子离子m/z为429.1186,二级碎片m/z有252.0428、251.0350、254.0585、132.0217、135.0452,保留时间为10.89min;甘草素准分子离子m/z为255.0657,二级碎片m/z为119.0502、135.0088、117.0346、213.0557、109.0295,保留时间为12.64min;甘草酸准分子离子m/z为821.3960,二级碎片m/z为351.1057、193.0354、759.3961、803.3859、175.0248,保留时间为16.51min;白玉兰亭b准分子离子m/z为355.1545,二级碎片m/z有323.1289、203.0714、283.0612、135.0452、254.0585,保留时间为19.74min;去甲脱水淫羊藿黄素准分子离子m/z为353.1025,二级碎片m/z有297.0405、284.0326、269.0456、281.0456227.0350,保留时间为20.58min;甘草利酮准分子离子m/z为381.1338,二级碎片m/z有279.0299、335.0925、366.1109、311.0561、308.0326,保留时间为21.90min;光甘草定准分子离子m/z为323.1283,二级碎片m/z有135.0452、323.1289、201.0921、253.0506、187.0764,保留时间为24.05min;砂生槐异黄酮a准分子离子m/z为351.0869,二级碎片m/z有333.0768、335.0561、199.0164、215.0714、203.0714,保留时间为25.55min;补骨脂定准分子离子m/z为335.0919,二级碎片m/z有319.0612、317.0819、293.0456、176.0115、161.0244,保留时间为27.46min;甘草查耳酮a准分子离子m/z为337.1440,二级碎片m/z有297.1132、307.0976、201.0921、149.0608,保留时间为32.00min;桑皮酮c准分子离子m/z为421.1651,二级碎片m/z有365.1031、407.1864、309.0405、337.1082、323.0561,保留时间为32.71min;甘草亭酸准分子离子m/z为469.3318,二级碎片m/z有201.0121、219.1027、425.3425、221.1183、205.0870,保留时间为33.69min;脱氧甘草亭酸准分子离子m/z为455.3525,二级碎片m/z有251.2380、135.0451、429.3374、161.0972、389.3061,保留时间为36.77min。筛查时应符合保留时间限定范围为
±
0.5min,精确质量偏差为5mda,离子化形式选择+h模式,匹配3个以上碎片信息,则确认该化合物。
12.有益效果:
13.本发明提供了一种uplc-q-tof-ms快速筛查甘草中活性成分的方法,可实现无标准品对照情况下20种活性成分的定性筛查,为甘草及其制品的产地溯源、真假鉴别和质量分级提供了一种新的有效方法,同时对市场监管和质量保证具有重要意义。
附图说明
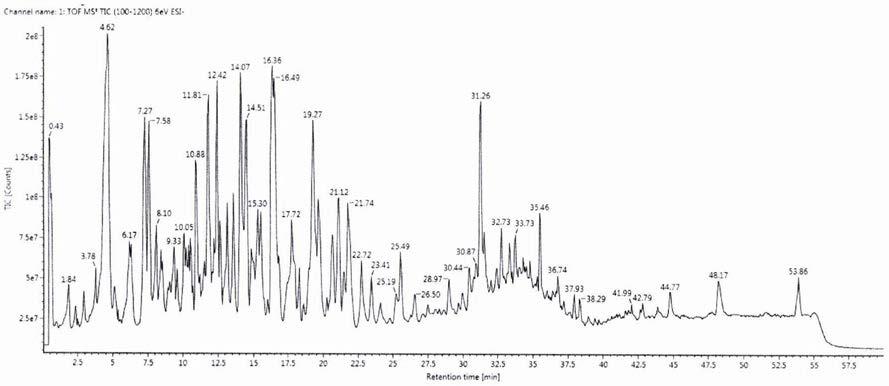
14.图1.甘草样品的总离子流图
15.图2.阿魏酸的提取离子色谱图和ms
e
模式下的高碰撞能通道质谱图及二级碎片匹配情况
16.图3.异甘草苷芹糖的提取离子色谱图和ms
e
模式下的高碰撞能通道质谱图及二级碎片匹配情况
17.图4.甘草苷的提取离子色谱图和ms
e
模式下的高碰撞能通道质谱图及二级碎片匹配情况
18.图5.甘草苷元的提取离子色谱图和ms
e
模式下的高碰撞能通道质谱图及二级碎片匹配情况
19.图6.新甘草苷的提取离子色谱图和ms
e
模式下的高碰撞能通道质谱图及二级碎片匹配情况
20.图7.异甘草素的提取离子色谱图和ms
e
模式下的高碰撞能通道质谱图及二级碎片匹配情况
21.图8.芒柄花素的提取离子色谱图和ms
e
模式下的高碰撞能通道质谱图及二级碎片匹配情况
22.图9.芒柄花苷的提取离子色谱图和ms
e
模式下的高碰撞能通道质谱图及二级碎片匹配情况
23.图10.甘草素的提取离子色谱图和ms
e
模式下的高碰撞能通道质谱图及二级碎片匹配情况
24.图11.甘草酸的提取离子色谱图和ms
e
模式下的高碰撞能通道质谱图及二级碎片匹配情况
25.图12.白玉兰亭b的提取离子色谱图和ms
e
模式下的高碰撞能通道质谱图及二级碎片匹配情况
26.图13.去甲脱水淫羊藿黄素的提取离子色谱图和ms
e
模式下的高碰撞能通道质谱图及二级碎片匹配情况
27.图14.甘草利酮的提取离子色谱图和ms
e
模式下的高碰撞能通道质谱图及二级碎片匹配情况
28.图15.光甘草定的提取离子色谱图和ms
e
模式下的高碰撞能通道质谱图及二级碎片匹配情况
29.图16.砂生槐异黄酮a的提取离子色谱图和ms
e
模式下的高碰撞能通道质谱图及二级碎片匹配情况
30.图17.补骨脂定的提取离子色谱图和ms
e
模式下的高碰撞能通道质谱图及二级碎片匹配情况
31.图18.甘草查耳酮a的提取离子色谱图和ms
e
模式下的高碰撞能通道质谱图及二级碎片匹配情况
32.图19.桑皮酮c的提取离子色谱图和ms
e
模式下的高碰撞能通道质谱图及二级碎片匹配情况
33.图20.甘草亭酸的提取离子色谱图和ms
e
模式下的高碰撞能通道质谱图及二级碎片匹配情况
34.图21.脱氧甘草亭酸的提取离子色谱图和ms
e
模式下的高碰撞能通道质谱图及二
级碎片匹配情况
具体实施方式
35.实施例1甘草中活性成分筛查
36.1 材料与方法
37.1.1 材料与仪器
38.甘草,购于中国食品药品检定研究院;乙腈、甲醇、甲酸lc-ms级,thermofisher scientific;标准品:甘草酸氨、甘草苷、甘草次酸等纯度均≥93%,中国药品生物制品检定研究所;尼龙滤膜0.22μm,美国agela technologies公司。
39.waters xevo g2-xs qtof/uplc系统 电喷雾离子源 美国waters公司;unifi 1.8软件系统 美国waters公司;vortex-genie 2涡旋震荡器 美国scientific in.d.ustries公司;millipore-q超纯水净化仪 美国millipore-q公司;ab135-s分析天平,感量0.01g/0.01mg 瑞士mettler toledo公司;kh-500 e超声清洗器 中国昆山禾创超声仪器有限公司。
40.1.2 实验方法
41.1.2.1 色谱条件
42.色谱柱acquity uplc hss t3,100mm
×
2.1mm,1.8μm;流动相a相为含0.1%甲酸水溶液,b相为乙腈,梯度洗脱;梯度洗脱的比例为0~10min,a为90%
→
70%;10~30min,a为70%
→
50%;30~40min,a为50%
→
10%;40~53min,a为10%;53~55min,a为10%
→
90%;55~60min,a为90%;流速0.5ml/min,进样量5μl,柱温45℃。
43.1.2.2 质谱条件
44.离子源esi,负离子模式;离子源温度100℃;脱溶剂气温度550℃;脱溶剂气流速600l/h;碰撞能量:低碰撞能量off,高碰撞能量20~40v;毛细管电压2.0kv;锥孔电压40ev;锥孔气流速50l/h;扫描模式为灵敏度模式,扫描范围m/z 100~1200;扫描时间0.2s;数据采集模式为mse模式。
45.1.2.3 样品前处理
46.将样品粉碎、过60目不锈钢筛网,称取约0.5g于离心管中,加入约50ml75%乙醇溶解,涡旋混匀;然后超声提取10min,取出后8000r/min离心5min,取上清液,过0.22μm有机滤膜,收集滤液待测。
47.2 结果与分析
48.ms
e
模式采集样品,结合unifi软件平台,利用自建谱库进行筛查。结果显示,甘草样品中含有甘草苷、甘草次酸等20种活性成分,不少于3个碎片匹配成功且质量误差在5mda以内,甘草中各活性成分的提取离子色谱图和ms
e
模式下的高碰撞能通道质谱图及二级碎片匹配情况,如图2~图21所示。
49.上述实施例仅是本发明的较佳实施例,而并非是对本发明作任何限制,凡是根据本发明技术实质对以上实施例所作的任何简单修改、变更以及等效变换,均仍属于本发明技术方案的保护范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1