一种质量可控的板蓝根提取物以及一种鉴别中成药中含有原料药材板蓝根的方法与流程
1.本发明属于中药提取物以及中成药的质量控制领域,具体涉及一种质量可控的板蓝根提取物以及一种鉴别中成药中含有原料药材板蓝根的方法。
背景技术:
2.板蓝根为十字花科植物菘蓝isatis indigotica fort.的干燥根,是一种历史悠久的传统中药,具有清热解毒、凉血、利咽等功效。自1977年以来,板蓝根在历版《中国药典》中均有收载,不仅以饮片形式在中医方剂中入药,也是多种中成药的工业原料。板蓝根化学成分极其复杂,过去数十年来,许多学者致力于板蓝根活性成分研究,从中分离出化合物多达约200个,涉及生物碱、有机酸、蒽醌、黄酮、木脂素、三萜、甾醇、芥子苷、核苷、脂肪酸、氨基酸、碳水化合物等结构类型。申请号为cn200510012767.7的中国专利申请公开了一种板蓝根提取物,它是由下列制备步骤制得:取板蓝根饮片,加50~85%乙醇溶液5~15倍量,置于合适容器中,密闭,冷浸;加热回流提取,过滤,减压回收乙醇,得药液;真空度控制在20~90kpa将药液减压浓缩至得到稠膏;将稠膏于50~80℃干燥,得到产物。但是,该提取物中成分复杂,质量可控性较差,该申请没有给出是否能够利用该提取物对含有板蓝根的中成药进行鉴别的方法。
3.风热感冒颗粒为临床常用的含有板蓝根的中成药,由板蓝根、连翘、芦根、牛蒡子、菊花、连翘等11味中药制成,具有清温解毒、宣肺利咽的作用,用于治疗感冒身热、鼻塞、头痛、咳嗽、多痰。风热感冒颗粒法定标准为部颁药品标准,即卫生部药品标准中药成方制剂第一册(ws3
‑
0044
‑
089)。从颁布至今仅对其处方、制法、性状作了规定要求,并按颗粒剂通则进行检验,无其他质量控制项目。目前对该品种只能按本标准的性状和《中国药典》2015年版四部中颗粒剂通则项下的内容进行检验。因此,为了进一步准确控制风热感冒颗粒的质量,需要建立更多的质量控制项目。
4.文献《风热感冒颗粒的薄层色谱鉴别,曾晓瑜等》记载了一种采用薄层色谱(tlc)法对该药的主要药材板蓝根、芦根、牛蒡子、连翘进行定性鉴别的方法。结果表明,利用薄层色谱法,以正丁醇一无水乙醇一氯仿一乙酸一水(5:5:2:1:1)为展开剂,能够对风热感冒颗粒中的板蓝根行定性鉴别。但是,文献末尾也记载了板蓝根药材中含有谷氨酸、脯氨酸、一氨基丁酸、精氨酸等,其中精氨酸的极性较强,当温度较低时,薄层色谱法展开剂的极性可能改变。因此利用该薄层色谱法定性鉴别风热感冒颗粒中的板蓝根时耐用性较差。此外,该文献记载的方法不是利用板蓝根特有的指标成分来进行鉴别的,因此不能有效的将风热感冒颗粒中的板蓝根与其它药材区别开,不利于风热感冒颗粒中板蓝根的鉴别。而且,薄层色谱法多使用有毒有害的有机试剂,对环境和人员都会造成一定程度的伤害。因此,亟需开发一种安全环保、耐用性好、专属性好、灵敏度高的方法来进一步对风热感冒颗粒中的原料药进行鉴别,为风热感冒颗粒质量标准的建立提供依据。
5.目前,还未见以质量可控的板蓝根提取物为对照,利用气相色谱法来对含有板蓝
根的中成药(例如风热感冒颗粒)中的板蓝根进行鉴别的报道。
技术实现要素:
6.本发明的目的在于提供一种质量可控的板蓝根提取物,本发明的另一个目的在于提供一种鉴别中成药中含有原料药材板蓝根的方法,为含有原料药材板蓝根的中成药的质量标准建立提供依据。
7.本发明提供了一种板蓝根提取物,所述板蓝根提取物采用气相色谱法检测,至少含有以下特征峰:峰1:保留时间为9.46
±
0.050min,峰2:保留时间为10.95
±
0.050min;
8.所述的气相色谱条件为:
9.色谱柱:db
‑
wax毛细管柱;
10.载气:氮气,载气流速1~3ml/min;
11.检测器:fid检测器,检测温度220~240℃;
12.进样口温度:220~240℃。
13.进一步地,所述气相色谱条件中,色谱柱为db
‑
wax毛细管柱,30m
×
0.32mm
×
0.50μm;所述载气流速为2ml/min;所述检测温度为230℃;所述进样口温度为230℃;
14.和/或,所述气相色谱条件中,进样量为0.5~5.0μl,优选为1μl;所述气相色谱条件中采用分流进样,分流比为1:1。
15.进一步地,所述气相色谱条件中,柱温为程序升温:初始温度50℃,然后以每分钟5℃升至140℃,保持4分钟;然后以每分钟20℃升至200℃,保持1分钟;再以每分钟50℃升至235℃,保持3分钟。
16.进一步地,所述板蓝根提取物的制备方法为:取板蓝根药材,加水煎煮,过滤,取滤液,加碱,水蒸气蒸馏,收集馏出液,然后在馏出液中加入有机溶剂提取,提取后静置,取有机相,即得板蓝根提取物。
17.进一步地,所述煎煮时间为1~4小时;所述板蓝根药材与水的质量体积比为1:(15~25)g/ml;所述碱为无机碱,优选为氢氧化钠;
18.和/或,所述有机溶剂为石油醚;所述馏出液与石油醚的体积比为(40~60):1;所述提取方式为振摇提取。
19.进一步地,所述煎煮时间为2小时;所述板蓝根药材与水的质量体积比为1:20g/ml;所述碱与滤液的质量体积比为0.05~0.40g/ml,优选为0.20g/ml;
20.和/或,所述石油醚为石油醚60~90℃;所述馏出液与石油醚的体积比为50:1。
21.本发明还提供了一种鉴别中成药中含有原料药材板蓝根的方法,所述鉴别方法包括以下步骤:
22.a、取中成药,制备成供试品溶液;
23.b、取上述的板蓝根提取物,制备成对照品溶液;
24.c、采用上述的气相色谱条件检测;
25.d、若供试品溶液的气相色谱图中至少含有以下特征峰:峰1保留时间为9.46
±
0.050min、峰2保留时间为10.95
±
0.050min,则中成药中含有板蓝根药材;若不含有以下特征峰或只含以下特征峰中的一个:峰1保留时间为9.46
±
0.050min、峰2保留时间为10.95
±
0.050min,则中成药中不含有板蓝根药材。
26.进一步地,步骤a中,所述供试品溶液的制备方法为:取中成药,加碱性水溶液,水蒸气蒸馏,收集馏出液,然后在馏出液中加入有机溶剂提取,提取后静置,取有机相,即得供试品溶液;
27.优选的,所述中成药是风热感冒颗粒。
28.进一步地,所述碱性水溶液为无机碱的水溶液,优选为氢氧化钠水溶液;所述中成药与碱性水溶液的质量体积比为0.5~2.0:1g/ml;
29.和/或,所述有机溶剂为石油醚;所述馏出液与石油醚的体积比为(40~60):1;所述提取方式为振摇提取。
30.进一步地,所述氢氧化钠水溶液的浓度为0.05~0.40g/ml,优选为0.20g/ml;所述中成药与碱性水溶液的质量体积比为1:1g/ml;
31.和/或,所述石油醚为石油醚60~90℃;所述馏出液与石油醚的体积比为50:1。
32.本发明中,石油醚(60~90℃)即石油醚60~90℃,是指石油醚中初馏点不低于60℃,终馏点不高于90℃的馏分。
33.本发明中,氢氧化钠溶液浓度是指质量体积浓度。例如,20%氢氧化钠溶液指溶液中氢氧化钠浓度为20%g/ml,即0.2g/ml。
34.本发明提供的板蓝根提取物质量可控,以该板蓝根提取物为对照品,利用本发明的气相色谱条件能够准确鉴别出风热感冒颗粒中的板蓝根药材,该方法安全环保、专属性好、耐用性好、选择性好、灵敏度高,为建立风热感冒颗粒的质量控制标准提供了支持。
35.利用本发明的方法不仅能够对风热感冒颗粒中的板蓝根药材进行定性鉴别,以主峰1、2对应的化合物2,4
‑
pentadienenitrile为对照品,还能定量测定风热感冒颗粒中的板蓝根药材的含量。
36.利用本发明的方法不仅能够对风热感冒颗粒中的板蓝根药材进行鉴别,还能对其它中成药中的板蓝根药材进行鉴别,为含有板蓝根药材的中成药的质量标准建立提供了依据。
37.显然,根据本发明的上述内容,按照本领域的普通技术知识和惯用手段,在不脱离本发明上述基本技术思想前提下,还可以做出其它多种形式的修改、替换或变更。
38.以下通过实施例形式的具体实施方式,对本发明的上述内容再作进一步的详细说明。但不应将此理解为本发明上述主题的范围仅限于以下的实例。凡基于本发明上述内容所实现的技术均属于本发明的范围。
附图说明
39.图1为对照药材溶液的气相色谱图。
40.图2为供试品溶液的气相色谱图。
41.图3为缺板蓝根阴性样品溶液的气相色谱图。
42.图4为采用a公司生产的风热感冒颗粒所得供试品溶液的气相色谱图。
43.图5为采用b公司生产的风热感冒颗粒所得供试品溶液的气相色谱图。
44.图6为采用c公司生产的风热感冒颗粒所得供试品溶液的气相色谱图。
45.图7为采用d公司生产的风热感冒颗粒所得供试品溶液的气相色谱图。
46.图8为采用e公司生产的风热感冒颗粒所得供试品溶液的气相色谱图。
47.图9为采用f公司生产的风热感冒颗粒所得供试品溶液的气相色谱图。
48.图10为采用g公司生产的风热感冒颗粒所得供试品溶液的气相色谱图。
49.图11为采用h公司生产的风热感冒颗粒所得供试品溶液的气相色谱图。
50.图12为采用i公司生产的风热感冒颗粒所得供试品溶液的气相色谱图。
51.图13为采用j公司生产的风热感冒颗粒所得供试品溶液的气相色谱图。
52.图14为采用k公司生产的风热感冒颗粒所得供试品溶液的气相色谱图。
53.图15为采用l公司生产的风热感冒颗粒所得供试品溶液的气相色谱图。
54.图16为采用m公司生产的风热感冒颗粒所得供试品溶液的气相色谱图。
55.图17为采用n公司生产的风热感冒颗粒所得供试品溶液的气相色谱图。
56.图18为采用o公司生产的风热感冒颗粒所得供试品溶液的气相色谱图。
57.图19为板蓝根对照药材溶液气质联用测得的总离子流谱图。
58.图20为主峰1对应化合物的离子质谱图。
59.图21为主峰1对应化合物的结构图。
60.图22为主峰2对应化合物的离子质谱图。
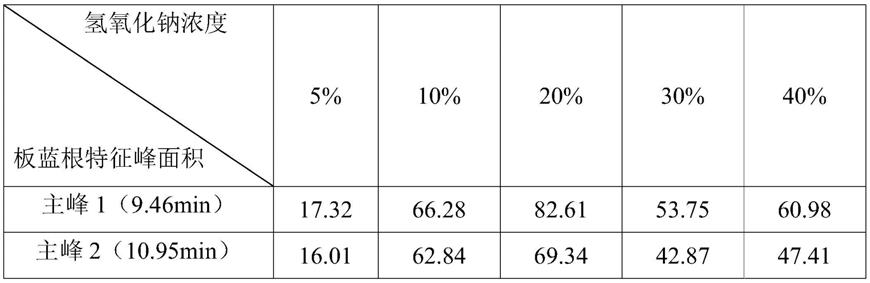
61.图23为主峰2对应化合物的结构图。
62.图24为表2中氢氧化钠溶液浓度为20%时对应的供试品溶液气相色谱图。
具体实施方式
63.本发明所用原料与设备均为已知产品,通过购买市售产品所得。
64.仪器:agilent 7890b气相色谱仪(安捷伦科技有限公司);xpe26电子天平(瑞士梅特勒
‑
托利多公司);ae
‑
240电子天平(德国赛多利斯公司);buchi distillation unit k
‑
350水蒸气蒸馏器。
65.板蓝根对照药材(121177
‑
201608)由中国食品药品检定研究院提供。
66.实施例1风热感冒颗粒中板蓝根的气相色谱鉴别方法
67.按照气相色谱法(《中国药典2020年版》四部通则0521)测试,具体步骤如下:
68.对照药材溶液的制备:取板蓝根对照药材5g,加水100ml煎煮2小时,滤过,得滤液50ml,置蒸馏管中,加入氢氧化钠10g,水蒸气蒸馏,收集馏出液250ml,置容量瓶(容量瓶中预先装入石油醚(60~90℃)5ml)中,振摇提取,摇匀后静置,取上层石油醚溶液,作为对照药材溶液。
69.供试品溶液的制备:取市售风热感冒颗粒50g,置蒸馏管中,加入20%氢氧化钠溶液50ml,水蒸气蒸馏,收集馏出液250ml,置容量瓶(容量瓶中预先装入石油醚(60~90℃)5ml)中,振摇提取,摇匀后静置,取上层石油醚溶液,作为供试品溶液。
70.测定法分别精密吸取对照药材溶液与供试品溶液各1μl,注入气相色谱仪,测定,即得。色谱条件如下:
71.使用db
‑
wax(聚乙二醇)毛细管柱(30m
×
0.32mm
×
0.50μm);fid检测器,温度230℃;进样口温度230℃;分流进样,分流比1:1;载气为氮气,载气流速:2ml/min;柱温为程序升温(见表1):初始温度50℃,以每分钟5℃升至140℃,保持4分钟,以每分钟20℃升至200℃,保持1分钟,再以每分钟50℃升至235℃,保持3分钟。理论板数按板蓝根主峰1计算,不得低于2
×
105。
72.表1柱温程序升温设置
73.升温速率(℃/分钟)柱温箱温度(℃)保持时间(分钟)
‑
50051404202001502353
74.结果判断结果如图1~2所示。本实施例采用水蒸气蒸馏法提取所得供试品溶液色谱图杂质少,峰形佳,干扰小。供试品溶液色谱图中出现与对照药材溶液色谱图中保留时间相同的2个主色谱峰(主峰1:9.46min、主峰2:10.95min),判断供试品风热感冒颗粒中含有板蓝根药材。
75.以下通过实验例证明本发明的有益效果。
76.实验例1色谱柱筛选实验
77.1、实验方法
78.参照实施例1的测试方法,将db
‑
wax(聚乙二醇)毛细管柱(30m
×
0.32mm
×
0.50μm)分别更换为db
‑
wax(聚乙二醇)毛细管柱(30m
×
0.53mm
×
1.0μm)或hp
‑
5毛细管柱(30m
×
0.32mm
×
0.25μm),其余检测条件不变,分析所得的色谱图。
79.2、实验结果
80.结果发现,采用db
‑
wax(聚乙二醇)毛细管柱(30m
×
0.32mm
×
0.50μm)时,理论塔板数高,分离度最好,保留时间适中。故选用db
‑
wax(聚乙二醇)毛细管柱(30m
×
0.32mm
×
0.50μm)。
81.实验例2供试品溶液制备方法的筛选
82.2.1振摇提取溶剂的筛选
83.参照实施例1的方法制备供试品溶液,仅仅将石油醚(60~90℃)更换为乙酸乙酯。但是发现振摇提取后静置,体系无法分层,溶剂混溶。
84.故选用石油醚(60~90℃)作为振摇提取溶剂。
85.2.2提取方法的筛选
86.参照实施例1的测试方法,仅仅将供试品溶液和对照药材溶液的制备方法修改为以下挥发油提取方法:
87.供试品溶液的制备方法:取风热感冒颗粒50g,置挥发油提取器中,连接挥发油提取器,加20%氢氧化钠溶液350ml,在挥发油提取器支管中加入石油醚(60~90℃)5ml,照挥发油测定法(《中国药典》2015年版四部通则2204)保持微沸3小时,分取石油醚液,定容至5ml,作为供试品溶液。
88.对照药材溶液的制备方法:取板蓝根对照药材5g,加水100ml煎煮2小时,滤过,得滤液50ml,置挥发油提取器中,加入氢氧化钠10g,在挥发油提取器支管中加入石油醚(60~90℃)5ml,照挥发油测定法(《中国药典》2015年版四部通则2204)保持微沸3小时,分取石油醚液,定容至5ml,作为对照药材溶液。
89.结果发现上述挥发油提取方法所得供试品溶液色谱图杂质多,峰形不清晰。故选用实施例1中的水蒸气蒸馏法来制备供试品溶液。
90.2.3不同浓度的碱对提取效果的考察
91.参照实施例1的测试方法,仅仅将供试品溶液的制备方法修改为:取参比制剂(由太极集团四川绵阳制药有限公司提供的风热感冒颗粒,批号200403)50g,分别加入5%、10%、20%、30%、40%的氢氧化钠溶液50ml,水蒸气蒸馏,收集馏出液250ml,置容量瓶(容量瓶中预先装入石油醚(60~90℃)5ml)中,振摇提取,摇匀后静置,取上层石油醚溶液,作为供试品溶液。
92.不同浓度的碱存在下所得供试品溶液检测所得气相色谱图中板蓝根特征峰面积对比结果见表2。氢氧化钠溶液浓度为20%时所得气相色谱图如图24所示。
93.表2不同浓度的碱对板蓝根鉴别的影响
[0094][0095]
由表2可知,在制备供试品溶液时,当氢氧化钠溶液浓度为20%时,所得色谱图中板蓝根特征峰面积最大。故在制备供试品溶液时选择20%氢氧化钠溶液。
[0096]
实验例3专属性试验
[0097]
采用与实施例1相同的方法制备对照药材溶液、供试品溶液。
[0098]
然后制备缺板蓝根阴性样品溶液:取风热感冒颗粒处方组成中除板蓝根外的其余药味制成缺板蓝根的阴性样品。然后参照实施例1供试品溶液的制备方法,仅仅将风热感冒颗粒替换为上述缺板蓝根的阴性样品,制得缺板蓝根阴性样品溶液。
[0099]
精密吸取对照药材溶液、供试品溶液、缺板蓝根阴性样品溶液各1μl,分别注入气相色谱仪进行测定。色谱条件与实施例1相同。
[0100]
结果如图1~3所示。供试品溶液色谱图中出现与对照药材溶液色谱图中保留时间相同的2个主色谱峰(主峰1:9.46min、主峰2:10.95min)。缺板蓝根阴性样品溶液则在对照药材溶液色谱图相应保留时间处无色谱峰。说明缺板蓝根阴性样品对板蓝根鉴别无干扰,本发明测试方法专属性好。
[0101]
实验例4耐用性的考察
[0102]
以不同厂家(a公司~o公司)生产的市售风热感冒颗粒为样品,参照实施例1的方法制备各供试品溶液,然后按照实施例1的测试方法得到各供试品溶液色谱图。
[0103]
结果如图4~18所示,可以看出,本发明的测试方法耐用性好。
[0104]
实验例5 2个主色谱峰对应的化合物结构确认
[0105]
按照实施例1的方法制备板蓝根对照药材溶液,然后通过气相色谱质谱联用进行成分确证(气相色谱条件与实施例1相同)。所得总离子流(tic)谱图如图19所示,离子质谱图如图20、22所示。
[0106]
分析后确认板蓝根对照药材溶液主峰1、2对应的化合物为2,4
‑
pentadienenitrile。两个主峰对应的化合物分子量、分子式相同,分子立体空间构象上存
在顺反式结构,推论主峰1、2为顺反异构体。主峰1对应化合物的nist ms search结构如图21所示,主峰1对应化合物的nist ms search结构如图23所示。
[0107]
因此,利用本发明的方法,以主峰1、2对应的化合物2,4
‑
pentadienenitrile为对照品,还能定量测定风热感冒颗粒中的板蓝根药材的含量。
[0108]
综上,本发明提供了一种板蓝根提取物以及一种鉴别中成药中含有原料药材板蓝根的方法。该板蓝根提取物质量可控,以该板蓝根提取物为对照品,利用本发明的气相色谱条件能够准确鉴别出风热感冒颗粒中的板蓝根药材,该方法安全环保、专属性好、耐用性好、选择性好、灵敏度高,为含有板蓝根药材的中成药的质量标准建立提供了依据。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1