一种枸橼酸马罗匹坦及其有关物质的检测方法与流程
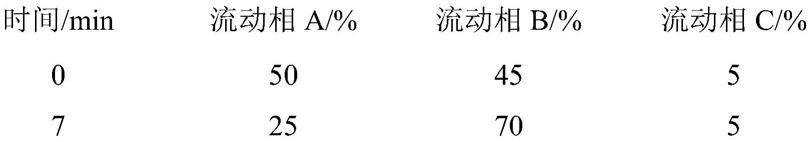
1.本发明涉及药物分析技术领域,尤其涉及一种枸橼酸马罗匹坦及其有关物质的检测方法。
背景技术:
2.枸橼酸马罗匹坦(英文名:maropitant citrate;cas号:147116
‑
67
‑
4)为神经激肽(nk1)受体拮抗剂,能够阻断p物质(与呕吐相关的神经递质)与受体的结合,可以有效抑制中枢神经性呕吐、外周神经性呕吐以及化学因素引起的呕吐,是第一种用于预防和治疗犬类严重呕吐和晕动病的药物,也是治疗犬晕动病首次获准上市的药物,疗效确切,具有吸收快,生物利用度高,药效持久且安全性高等优点,具有良好的临床应用价值和市场前景。
3.枸橼酸马罗匹坦合成过程中可能会引入起始原料、中间体、副产物等杂质,所以必须建立相应的检测方法从而控制杂质的含量来保证用药的安全。但目前,《美国药典》、《欧洲药典》、《日本药典》及《中国药典》中并未收录枸橼酸马罗匹坦有关物质的检测方法,也未检测到有关文献报道枸橼酸马罗匹坦有关物质的检测方法。但有关物质对于临床应用的安全性可能存在一定的不良影响,因此,对枸橼酸马罗匹坦进行质量控制就显得尤为重要。
技术实现要素:
4.针对现有技术中尚无枸橼酸马罗匹坦有关物质的检测方法的问题,本发明提供一种枸橼酸马罗匹坦及其有关物质的检测方法。
5.为达到上述发明目的,本发明实施例采用了如下的技术方案:
6.一种枸橼酸马罗匹坦及其有关物质的检测方法,以0.1~0.2%v/v三乙胺的甲醇溶液为溶剂配制待测溶液,用高效液相色谱法进行有关物质的测定,所述高效液相色谱法的色谱条件为:
7.色谱柱:十八烷基键合硅胶柱;
8.流动相:流动相a为0.005~0.015mol/l的磷酸氢二钾溶液,用磷酸调节ph值至7.30~7.70;流动相b为乙腈;流动相c为甲醇;
9.采用流动相a、流动相b和流动相c进行梯度洗脱,所述洗脱梯度如下:
10.11.检测波长为200~240nm;
12.柱温为38~42℃。
13.本发明所提供的枸橼酸马罗匹坦及其有关物质的检测方法能够实现主成分与已知杂质之间的有效分离,准确定性及定量检测枸橼酸马罗匹坦及其杂质情况,且经专属性、灵敏度等方法学研究及验证发现,本发明的检测方法色谱峰分离效果好,灵敏度高,准确度和重现性较好,且方法简单,操作方便,填补了枸橼酸马罗匹坦有关物质检测领域的空白,为提高和更好地控制枸橼酸马罗匹坦产品的质量提供了可靠保障,具有广阔的应用前景。
14.以流动相a为例,该洗脱梯度具体表示为:0min~7min,流动相a的体积百分比由49~51%匀速降至25%,7min~9min流动相a的体积百分比由25%匀速降至21.5%,30min~30.1min流动相a的体积百分比由21.5%匀速升至49~51%,之后一直为49~51%
15.优选地,所述有关物质包括2
‑
苯甲叉基奎宁酮、(2s)
‑2‑
二苯甲基奎宁酮、(2s,3s)
‑2‑
二苯甲基奎宁酮苄胺、(2s,3s)
‑2‑
二苯甲基奎宁酮
‑3‑
胺、5
‑
叔丁基
‑
2甲氧基苯甲醛和(2s,3s)
‑2‑
二苯甲基
‑
n
‑
(5
‑
叔丁基
‑2‑
甲氧基苄基)奎宁环
‑3‑
亚胺。以上杂质为枸橼酸马罗匹坦合成工艺中引入的或降解生成的杂质。
16.优选地,所述洗脱梯度如下:
[0017][0018]
优选地,所述流速为1.0ml/min。
[0019]
优选地,所述检测波长为222nm。
[0020]
优选地,所述柱温为40℃。
[0021]
优选地,所述流动相a的ph值为7.50。
[0022]
优选地,所述磷酸氢二钾溶液中磷酸氢二钾的浓度为0.01mol/l。
[0023]
优选地,以0.2%v/v三乙胺的甲醇溶液为溶剂配制待测溶液。
[0024]
优选地,所述色谱柱为capcell pak c18,规格为:4.6
×
250mm,填料粒径5μm。
[0025]
优选地,该检测方法包括以下步骤:
[0026]
步骤a、配制供试品溶液和枸橼酸马罗匹坦及其有关物质的至少五个浓度的对照品溶液;
[0027]
步骤b、用所述高效液相色谱法对所述对照品溶液进行测定,得到线性回归方程;
[0028]
步骤c、用所述高效液相色谱法对所述供试品溶液进行测定,利用步骤b所得线性回归方程计算所述供试品溶液中枸橼酸马罗匹坦及其有关物质的含量。
[0029]
本发明的有益效果在于:色谱条件专属性高,系统稳定,线性好,精密度高,准确度高,耐用性试验良好,可有效测定枸橼酸马罗匹坦及其有关物质。
附图说明
[0030]
图1为实施例1中系统适用性项下系统适用性溶液的色谱图;
[0031]
图2为实施例1中专属性项下供试品溶液的色谱图;
[0032]
图3为实施例1中检测限定量限项下检测限溶液的色谱图;
[0033]
图4为实施例1中检测限定量限项下定量限溶液的色谱图。
具体实施方式
[0034]
为了使本发明的目的、技术方案及优点更加清楚明白,以下结合附图及实施例,对本发明进行进一步详细说明。应当理解,此处所描述的具体实施例仅仅用以解释本发明,并不用于限定本发明。
[0035]
在以下实施例中,杂质a是指2
‑
苯甲叉基奎宁酮,杂质b是指(2s)
‑2‑
二苯甲基奎宁酮,杂质c是(2s,3s)
‑2‑
二苯甲基奎宁酮苄胺,杂质d是指(2s,3s)
‑2‑
二苯甲基奎宁酮
‑3‑
胺,杂质e是指5
‑
叔丁基
‑
2甲氧基苯甲醛,杂质f是指(2s,3s)
‑2‑
二苯甲基
‑
n
‑
(5
‑
叔丁基
‑2‑
甲氧基苄基)奎宁环
‑3‑
亚胺。
[0036]
仪器:高效液相色谱仪,二极管阵列检测器,量瓶,电子天平。
[0037]
空白溶剂:0.2%v/v三乙胺的甲醇溶液。
[0038]
实施例1
[0039]
本发明实施例提供了一种枸橼酸马罗匹坦及其有关物质的检测方法。
[0040]
1.1溶液的配制
[0041]
各杂质储备液的配制:分别取杂质a、杂质b、杂质c、杂质d、杂质e、杂质f对照品适量,精密称定,分别加0.2%v/v三乙胺的甲醇溶液溶解并稀释制成1ml中约含0.08mg的各杂质储备液。
[0042]
供试品溶液的制备:取枸橼酸马罗匹坦供试品适量,加0.2%v/v三乙胺的甲醇溶液溶解并稀释制成1ml中含有枸橼酸马罗匹坦0.4mg/ml的供试品。
[0043]
对照品一级储备液的配制:精密量取各对照品2.0ml,置10ml的容量瓶内,用0.2%v/v三乙胺的甲醇溶液溶解定容至刻度,摇匀,作为各对照品一级储备液。
[0044]
混合对照品溶液:分别精密量取枸橼酸马罗匹坦对照品一级储备液、杂质a、杂质b、杂质c、杂质d、杂质e、杂质f杂质储备液适量,用0.2%v/v三乙胺的甲醇溶液稀释制成含有枸橼酸马罗匹坦、杂质a、杂质b、杂质c、杂质d、杂质e、杂质f含量均为0.8μg/ml的混合溶液。
[0045]
各杂质及枸橼酸马罗匹坦定位溶液的配制:分别精密量取枸橼酸马罗匹坦对照品一级储备液、杂质a、杂质b、杂质c、杂质d、杂质e、杂质f各杂质储备液适量,分别用0.2%v/v三乙胺的甲醇溶液稀释制成含有枸橼酸马罗匹坦、杂质a、杂质b、杂质c、杂质d、杂质e、杂质f含量为0.8μg/ml的定位溶液。
[0046]
1.2高效液相色谱的条件:
[0047]
色谱柱:capcell pak c18(4.6
×
250mm)5μm;
[0048]
流速:1.0ml/min;
[0049]
检测波长:222nm;
[0050]
柱温:40℃;
[0051]
供试品溶液浓度:0.4mg/ml;
[0052]
进样量:20μl;
[0053]
流动相a相:0.01mol/l的磷酸氢二钾溶液(ph7.50),配制方法为:取磷酸氢二钾2.28g,加水900ml使溶解后,用磷酸调节ph至7.50,加水至1000ml,摇匀,滤过,即得;
[0054]
流动相b相:乙腈;
[0055]
流动相c相:甲醇。
[0056]
按下列梯度程序进行洗脱:
[0057]
时间(min)流动相a(%)流动相b(%)流动相c(%)050455725705921.573.553021.573.5530.1504555050455
[0058]
1.3方法学验证:
[0059]
1、系统适用性
[0060]
取空白溶剂、混合对照品溶液(即系统适用性溶液)进样检测,其中系统适用性溶液连续进样6针,按照1.2的高效液相色谱条件进样检测,进样量20μl,记录色谱图,如图1所示。试验结果数据如表1所示。
[0061]
表1系统适用性试验结果
[0062][0063][0064]
由图1可见,基线平稳无干扰,空白溶剂对杂质及主成分检测无干扰;由表1数据可见,杂质a、b、c、d、e、f和枸橼酸马罗匹坦的峰面积与保留时间的rsd值<2%,均符合要求,且各杂质与主峰枸橼酸马罗匹坦的分离度均符合规定,适合有关物质的检查。
[0065]
该试验结果说明本方法的系统适用性良好。
[0066]
2、专属性
[0067]
取空白溶剂、杂质定位溶液、供试品溶液、混合对照品溶液分别按照1.2的高效液相色谱条件进样检测,进样量20μl,记录色谱图,供试品溶液的色谱图如图2所示,供试品溶液的试验结果数据如表2所示,混合对照溶液的试验结果数据如表3所示。
[0068]
表2供试品溶液试验结果
[0069]
名称保留时间(min)峰面积分离度杂质b8.0309787
‑
杂质c13.9402638613.267杂质e10.392216011.265枸橼酸马罗匹坦25.6371752397423.889
[0070]
供试品中杂质a、d、f未检出。
[0071]
表3混合对照品溶液的试验结果
[0072][0073][0074]
试验结果表明,各杂质峰之间、各杂质峰与枸橼酸马罗匹坦之间最小分离度均大于1.5,各杂质之间、各杂质与枸橼酸马罗匹坦之间无干扰,说明本方法专属性良好。
[0075]
3、检测限和定量限
[0076]
取各杂质储备液及枸橼酸马罗匹坦的对照品一级储备液溶液,用稀释法分别测其定量限(s/n≥10)、检测限(s/n≥3),检测限溶液的色谱图如图3所示,定量限溶液的色谱图如图4所示。试验结果数据见表4。
[0077]
表4检测限和定量限试验结果
[0078]
样品定量限浓度(μg/ml)检测限浓度(μg/ml)杂质a0.0960.032杂质b0.1000.033杂质c0.1850.061杂质d0.1600.053杂质e0.0400.013杂质f0.2950.097马罗匹坦0.3200.106
[0079]
试验结果表明,杂质a、b、c、d、e、f和枸橼酸马罗匹坦的检测限浓度分别是0.096μg/ml、0.100μg/ml、0.185μg/ml、0.160μg/ml、0.040μg/ml、0.295μg/ml、0.320μg/ml;定量限浓度分别是0.032μg/ml、0.033μg/ml、0.061μg/ml、0.053μg/ml、0.013μg/ml、0.097μg/ml、0.106μg/ml。该试验结果说明本方法灵敏度高。
[0080]
4、线性范围
[0081]
精密称取杂质a、杂质b、杂质c、杂质d、杂质e以及杂质f适量,分别用空白溶剂溶解并稀释制成浓度为1.60μg/ml、1.20μg/ml、0.96μg/ml、0.80μg/ml、0.64μg/ml、0.40μg/ml的一系列浓度溶液。
[0082]
精密称取枸橼酸马罗匹坦对照品适量,分别用空白溶剂溶解并稀释制成浓度为2.320μg/ml、1.740μg/ml、1.392μg/ml、1.160μg/ml、0.928μg/ml、0.580μg/ml的一系列浓度溶液。
[0083]
精密量取上述对照品溶液各20μl分别注入高效液相色谱仪,按照1.2的高效液相
色谱条件进样检测,记录色谱图,测定峰面积,以峰面积a为纵坐标、浓度c为横坐标作线性回归,结果见表5。
[0084]
表5线性结果
[0085]
样品线性方程r2线性范围(μg/ml)杂质ay=40569x+880.950.99970.40~1.60杂质by=38108x+431.930.99990.40~1.60杂质cy=36102x+32.4930.99960.40~1.60杂质dy=25049x
‑
122.540.99920.40~1.60杂质ey=102379x+3325.80.99980.40~1.60杂质fy=57808x
‑
2403.40.99960.40~1.60枸橼酸马罗匹坦y=33020x+13810.99950.58~2.32
[0086]
以上结果表明,杂质a、杂质b、杂质c、杂质d、杂质e以及杂质f在浓度为0.40~1.60μg/ml范围内r2分别为0.9997、0.9999、0.9996、0.9992、0.9998和0.9996,枸橼酸马罗匹坦在浓度为0.58~2.32μg/ml范围内r2=0.9995,说明本方法浓度和峰面积之间线性关系良好。
[0087]
5、准确度
[0088]
50%供试品溶液:称取29mg枸橼酸马罗匹坦加0.2%v/v三乙胺的甲醇溶液溶解,分别取杂质a、杂质b、杂质c、杂质d、杂质e、杂质f的对照品一级储备液适量,加0.2%v/v三乙胺的甲醇溶液制成含有杂质a、杂质b、杂质c、杂质d、杂质e、杂质f各0.4μg/ml、枸橼酸马罗匹坦0.4mg/ml的溶液,即得。
[0089]
100%供试品溶液:称取29mg枸橼酸马罗匹坦加稀释剂溶解,分别取杂质a、杂质b、杂质c、杂质d、杂质e、杂质f的对照品一级储备液适量,加0.2%v/v三乙胺的甲醇溶液制成含有杂质a、杂质b、杂质c、杂质d、杂质e、杂质f各0.8μg/ml、枸橼酸马罗匹坦0.4mg/ml的溶液,即得。
[0090]
150%供试品溶液:称取29mg枸橼酸马罗匹坦加稀释剂溶解,分别取杂质a、杂质b、杂质c、杂质d、杂质e、杂质f的对照品一级储备液适量,加0.2%v/v三乙胺的甲醇溶液制成含有杂质a、杂质b、杂质c、杂质d、杂质e、杂质f各1.2μg/ml、枸橼酸马罗匹坦0.4mg/ml的溶液,即得。
[0091]
照上述配制方法,每个浓度平行配制三份样品进行测定。
[0092]
精密量取上述溶液各20μl,注入液相色谱仪,按照1.2的高效液相色谱条件进样检测,记录色谱图,结果见下表6。
[0093]
表6回收率试验结果
[0094][0095]
以上结果表明,各杂质在各浓度下的回收率均在92~105%范围内,rsd值均小于2%,说明本方法准确度较好。
[0096]
实施例2
[0097]
本实施例提供一种枸橼酸马罗匹坦及其有关物质的检测方法,高效液相色谱的条件:
[0098]
色谱柱:capcell pak c18(4.6
×
250mm)5μm;
[0099]
流速:1.0ml/min;
[0100]
检测波长:222nm;
[0101]
柱温,38℃;
[0102]
供试品溶液浓度:0.4mg/ml;
[0103]
进样量:20μl;
[0104]
流动相a相:0.01mol/l磷酸氢二钾溶液(ph7.50),配制方法同实施例1;
[0105]
流动相b相:乙腈;
[0106]
流动相c相:甲醇。
[0107]
梯度程序条件与实施例1相同。
[0108]
精密量取空白溶剂以及实施例1的混合对照溶液、供试品溶液各20μl,分别注入液相色谱仪,按上述高效液相色谱条件进样测定,记录各成分峰面积记录色谱图,结果见表7、表8。
[0109]
表7混合对照溶液检测结果
[0110]
混合对照溶液保留时间min分离度杂质a9.3196.149杂质b8.13118.37杂质c13.85312.394杂质d4.614
‑
杂质e10.566.076杂质f28.974.346枸橼酸马罗匹坦25.81123.338
[0111]
表8供试品溶液有关物质检测结果
[0112]
供试品溶液保留时间min分离度杂质b8.128—
杂质c13.86211.610杂质e10.57611.146枸橼酸马罗匹坦25.77823.412
[0113]
实施例3
[0114]
本实施例提供一种枸橼酸马罗匹坦及其有关物质的检测方法,高效液相色谱的条件:
[0115]
色谱柱:capcell pak c18(4.6
×
250mm)5μm;
[0116]
流速:1.0ml/min;
[0117]
检测波长:222nm;
[0118]
柱温,42℃;
[0119]
供试品溶液浓度:0.4mg/ml;
[0120]
进样量:20μl;
[0121]
流动相a相:0.01mol/l磷酸氢二钾溶液(ph7.50),配制方法同实施例1;
[0122]
流动相b相:乙腈;
[0123]
流动相c相:甲醇。
[0124]
梯度洗脱条件与实施例1相同。
[0125]
精密量取空白溶剂以及实施例1的混合对照溶液、供试品溶液各20μl,分别注入液相色谱仪,按上述高效液相色谱条件进样测定,记录各成分峰面积记录色谱图,结果见表9、表10。
[0126]
表9混合对照溶液检测结果
[0127]
混合对照溶液保留时间min分离度杂质a9.1635.089杂质b8.06414.889杂质c13.96512.087杂质d4.803—杂质e10.3675.245杂质f28.4323.846枸橼酸马罗匹坦25.54321.273
[0128]
表10供试品溶液有关物质检测结果
[0129]
供试品溶液保留时间min分离度杂质b8.050—杂质c13.96211.064杂质e10.3708.499枸橼酸马罗匹坦25.50520.177
[0130]
实施例4
[0131]
本实施例提供一种枸橼酸马罗匹坦及其有关物质的检测方法,高效液相色谱的条件:
[0132]
色谱柱:capcell pak c18(4.6
×
250mm)5μm;
[0133]
流速:1.0ml/min;
[0134]
检测波长:222nm;
[0135]
柱温,40℃;
[0136]
供试品溶液浓度:0.4mg/ml;
[0137]
进样量:20μl;
[0138]
流动相a相:0.01mol/l磷酸氢二钾溶液(ph7.50),配制方法同实施例1;
[0139]
流动相b相:乙腈,
[0140]
流动相c相:甲醇。
[0141]
按下列梯度程序进行洗脱:
[0142]
时间(min)流动相a(%)流动相b(%)流动相c(%)049465725705921.573.553021.573.5530.1494655049465
[0143]
精密量取空白溶剂以及实施例1的混合对照溶液、供试品溶液各20μl,分别注入液相色谱仪,按上述高效液相色谱条件进样测定,记录各成分峰面积记录色谱图,结果见表11、表12。
[0144]
表11混合对照溶液检测结果
[0145][0146][0147]
表12供试品溶液有关物质检测结果
[0148]
供试品溶液保留时间min分离度杂质b7.963
‑
杂质c13.79215.008杂质e10.41712.717枸橼酸马罗匹坦25.68928.585
[0149]
实施例5
[0150]
本实施例提供一种枸橼酸马罗匹坦及其有关物质的检测方法,高效液相色谱的条
件:
[0151]
色谱柱:capcell pak c18(4.6
×
250mm)5μm;
[0152]
流速:1.0ml/min;
[0153]
检测波长:222nm;
[0154]
柱温,40℃;
[0155]
供试品溶液浓度:0.4mg/ml;
[0156]
进样量:20μl;
[0157]
流动相a相:0.01mol/l磷酸氢二钾溶液(ph7.50),配制方法同实施例1;
[0158]
流动相b相:乙腈;
[0159]
流动相c相:甲醇。
[0160]
按下列梯度程序进行洗脱:
[0161]
时间(min)流动相a(%)流动相b(%)流动相c(%)051445725705921.573.553021.573.5530.1514455051445
[0162]
精密量取空白溶剂以及实施例1的混合对照溶液、供试品溶液各20μl,分别注入液相色谱仪,按上述高效液相色谱条件进样测定,记录各成分峰面积记录色谱图,结果见表13、表14。
[0163]
表13混合对照溶液检测结果
[0164]
混合对照溶液保留时间min分离度杂质a9.3696.672杂质b8.24021.676杂质c13.95714.791杂质d4.621
‑
杂质e10.5936.998杂质f29.0085.101枸橼酸马罗匹坦25.88127.830
[0165]
表14供试品溶液有关物质检测结果
[0166]
供试品溶液保留时间min分离度杂质b8.193—杂质c13.95315.058杂质e10.58412.587枸橼酸马罗匹坦25.83628.439
[0167]
以上所述仅为本发明的较佳实施例而已,并不用以限制本发明,凡在本发明的精神和原则之内所作的任何修改、等同替换和改进等,均应包含在本发明的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1