一种测定水中甲基萘化合物的方法与流程
1.本发明涉及甲基萘测定技术领域,更具体地说是一种测定水中甲基萘化合物的方法。
背景技术:
2.甲基萘有2种同分异构体,即1
‑
甲基萘(1
‑
mn)和2
‑
甲基萘(2
‑
mn),或称为α
‑
甲基萘和β
‑
甲基萘。混合甲基萘(mmn)及其2种同分异构体都有广泛的用途。作为有机合成的原料,主要用于合成聚合物单体、印染载体、热载体、涂料、橡胶、增塑剂等,在市场上有广泛的应用,在化工、医药、印染等行业及企业日常生产用的溶剂油,导热油里有存在。2
‑
甲基萘多用于有机合成,也用于制维生素k和杀虫剂等。
3.甲基萘有异味,在水中的阈值低,属于低毒类物质,进入地表水会对水体产生污染,人体摄入后会对健康产生危害。对人体具有刺激作用,引发高浓度致溶血性贫血及肝、肾损害。亦可发生视神经炎和视网膜炎。重者可发生中毒性脑病和肝损害。口服中毒主要引起溶血和肝、肾损害,甚至发生急性肾功能衰竭和肝坏死。慢性中毒可引起头痛、乏力、恶心、呕吐和血液系统损害,白内障、视神经炎和视网膜病变,皮肤接触可引起皮炎等症状。各国环保、卫生部门已将甲基萘作为重要的检测指标之一,我国也早已将其列入优先监测的污染物。因此,对其灵敏快速的检测至关重要。
4.目前我国甲基萘的测定标准只有中华人民共和国黑色冶金工业标准《工业甲基萘中甲基萘、萘含量的气相色谱测定法》(yb/t 5154
‑
93)。国内外均尚无相应的标准方法用于测定水中甲基萘。本方法的制定对我国地下水中甲基萘的调查与评价具有重大意义,可用本方法对地下水中2种甲基萘进行调查与评价,目前已应用于河北省重点企业行业用地地下水样品的检测中。
技术实现要素:
5.为了克服现有技术的上述缺陷,本发明提供一种测定水中甲基萘化合物的方法。
6.为实现上述目的,本发明提供如下技术方案:
7.一种测定水中甲基萘化合物的方法,采用液液萃取法萃取样品中的甲基萘,萃取液经脱水、浓缩、净化和定容后经气相色谱
‑
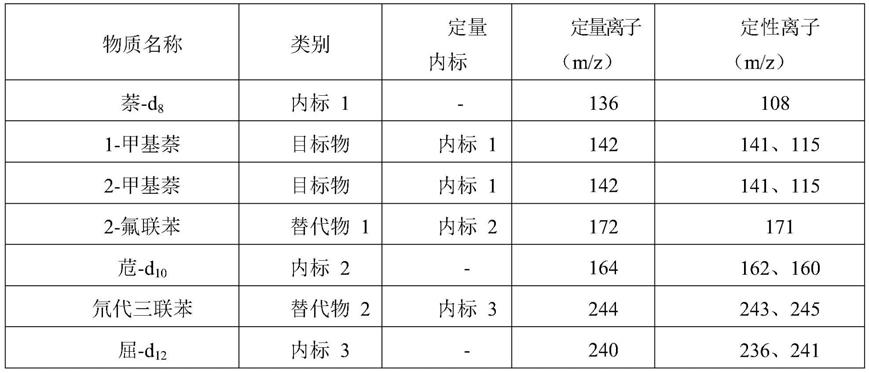
质谱法分离和测定。根据保留时间、碎片离子质荷比及不同离子丰度比定性,内标法定量。
8.具体步骤为:
9.一、试样提取
10.(1)采集与保存
11.样品应收集在棕色玻璃样品瓶中,水样充满样品瓶。在4℃下避光保存,14d内完成萃取。
12.(2)萃取
13.摇匀并准确量取水样1l至2l分液漏斗中,称取30g氯化钠加入到水样中,轻轻振摇
使其溶解。加入50ml二氯甲烷,加入替代物标准使用液,振摇10min。静置5min分层后,收集有机相,放入接收瓶中。重复萃取两次,合并有机相;萃取液中加入适量无水硫酸钠除水,稍稍摇动后放置20min以上,氮吹浓缩至1ml。
14.(3)净化
15.弗罗里硅土小柱用正己烷10ml活化后,在液面消失前,将上述预处理溶液加入到弗罗里硅土小柱上,用5ml正己烷洗涤浓缩管,洗涤液一并转移至弗罗里硅土小柱里,弃去流出液,用10ml二氯甲烷:正己烷(9:1)洗脱样品,收集于接收管中。
16.(4)浓缩
17.萃取液转移至氮吹管中,采用氮吹仪浓缩萃取液。氮吹浓缩仪设置温度30℃,压力设为1,小流量氮气将提取液浓缩至约1ml,用二氯甲烷定容至1.0ml,在上述定容后的溶液中加入内标标准使用溶液,摇匀,待测。制备的样品在4℃以下冷藏保存,30d内完成分析。
18.(5)空白试样的制备
19.用实验用水代替样品,按照试样制备相同的操作步骤,制备空白试样。
20.二、分析样品
21.(1)仪器参考条件
22.a气相色谱条件
23.程序升温:进样方式:不分流进样0.75min;进样量:1.0μl;进样口温度:250℃;柱流量:1.0ml/min。
24.b质谱参考条件
25.离子源温度:230℃;传输线温度:280℃;离子化能量:70ev;
26.全扫描(scan)质量范围:50
‑
500amu;
27.选择离子(sim)扫描,目标化合物扫描离子见表2。
28.表2目标化合物对应的扫描离子
[0029][0030]
(2)校准
[0031]
a仪器性能检查
[0032]
仪器使用前用全氟三丁胺对质谱仪进行调谐。样品分析前以及每运12h,将1.0μl十氟三苯基膦(dftpp)使用液注入色谱,对仪器系统进行检查,所得质量离子丰度应全部符
合表1中的要求。
[0033]
表1十氟三苯基膦(dftpp)关键离子及丰度标准
[0034]
质量离子丰度标准质量离子丰度标准68小于69峰的2%275基峰的10%~30%70小于69峰的2%365大于基峰的1%127基峰的40%~60%441存在且小于443峰197小于基峰的%442大于基峰的40%198基峰,丰度为100%443442峰的17%~23%
[0035]
(3)校准曲线的绘制
[0036]
分别吸取不同体积的标准和替代物标准使用液,配制成浓度为0.2、0.5、1.0、1.5、2.0μg/ml的标准系列,并同时加入内标使用液,混匀。按照仪器参考条件进行分析,得到不同目标化合物质谱图。以目标化合物浓度与内标化合物浓度的比值为横坐标,以目标化合物定量离子的响应值与内标化合物定量离子的响应值的比值为纵坐标,绘制校准曲线。
[0037]
(4)样品测定
[0038]
取待测试样按照与绘制校准曲线相同的仪器分析条件进行测定。
[0039]
(5)实验室空白试验
[0040]
在分析样品的同时,将空白试样按照与绘制校准曲线相同的仪器分析条件进行测定。
[0041]
三、结果计算与表示
[0042]
(1)定性分析
[0043]
以全扫描方式(scan)采集数据,以样品中目标化合物相对保留时间(rrt)、辅助定性离子和目标离子丰度比(q)与标准溶液中的变化范围来定性。样品中目标化合物的相对保留时间与校准曲线该化合物的平均相对保留时间的差值应在
±
0.06内。样品中目标化合物的辅助定性离子和定量离子峰面积比(q样品)与标准曲线目标化合物的辅助定性离子和定量离子峰面积比(q标准)相对偏差控制在
±
30%以内。
[0044]
按公式(1)计算相对保留时间rrt
[0045][0046]
式中:
[0047]
rt
c
—目标化合物的保留时间,min;
[0048]
rt
is
—内标物的保留时间,min。
[0049]
平均相对保留时间(rrt):标准系列中同一目标化合物的相对保留时间平均值
[0050]
按公式(2)计算辅助定性离子和定量离子峰面积比(q)
[0051][0052]
式中:
[0053]
a
t
—定量离子峰面积;
[0054]
a
q
—辅助定性离子峰面积。
[0055]
(2)定量分析
[0056]
以选择离子扫描方式(sim)采集数据,内标法定量。样品中目标物的质量浓度ρi(μg/l)按照公式(3)进行计算。
[0057][0058]
式中:
[0059]
ρ
i
—样品中甲基萘化合物或替代物的浓度,μg/l;
[0060]
ρ
is
—根据标准曲线查得甲基萘化合物或替代物的浓度,μg/ml;
[0061]
v—试样体积,ml;
[0062]
vt—水样体积,ml。
[0063]
通过大批量企业用地污染状况详查项目地下水样品的应用,此方法有以下优点:
[0064]
(1)样品提取稳定,平行性好,精密度高。
[0065]
(2)液液萃取操作简单,大多实验室都可以实现,可用于大批量检测工作。
[0066]
(3)检出限低,可定性定量精确的检测水中甲基萘。
[0067]
(4)样品加标和替代物回收率均能在70%~130%之间。
附图说明
[0068]
图1为本甲基萘标准物质的选择离子扫描总离子流图。
[0069]
图中化合物按保留时间排列依次为:
[0070]
1、2
‑
甲基萘;2、1
‑
甲基萘4;3、2
‑
氟联苯;4、氘代三联苯。
具体实施方式
[0071]
下面将对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
[0072]
一种测定水中甲基萘化合物的方法,采用液液萃取法萃取样品中的甲基萘,萃取液经脱水、浓缩、净化和定容后经气相色谱
‑
质谱法分离和测定。根据保留时间、碎片离子质荷比及不同离子丰度比定性,内标法定量。
[0073]
当取样量为1l时,本方法的检出限详见表1。
[0074]
表1目标化合物的名称及检出限和测定下限
[0075][0076]
具体实施方式如下:
[0077]
一、试样提取
[0078]
(1)采集与保存
[0079]
样品应收集在棕色玻璃样品瓶中,水样充满样品瓶。在4℃下避光保存,14d内完成萃取。
[0080]
(2)萃取
[0081]
摇匀并准确量取水样1l至2l分液漏斗中,称取30g氯化钠加入到水样中,轻轻振摇使其溶解。加入50ml二氯甲烷,加入替代物标准使用液,振摇10min。静置5min分层后,收集有机相,放入接收瓶中。重复萃取两次,合并有机相;萃取液中加入适量无水硫酸钠除水,稍稍摇动后放置20min以上,氮吹浓缩至1ml。
[0082]
(3)净化
[0083]
弗罗里硅土小柱用正己烷10ml活化后,在液面消失前,将上述预处理溶液加入到弗罗里硅土小柱上,用5ml正己烷洗涤浓缩管,洗涤液一并转移至弗罗里硅土小柱里,弃去流出液,用10ml二氯甲烷:正己烷(9:1)洗脱样品,收集于接收管中。
[0084]
(4)浓缩
[0085]
萃取液转移至氮吹管中,采用氮吹仪浓缩萃取液。氮吹浓缩仪设置温度30℃,压力设为1,小流量氮气将提取液浓缩至约1ml,用二氯甲烷定容至1.0ml,在上述定容后的溶液中加入内标标准使用溶液,摇匀,待测。制备的样品在4℃以下冷藏保存,30d内完成分析。
[0086]
(5)空白试样的制备
[0087]
用实验用水代替样品,按照试样制备相同的操作步骤,制备空白试样。
[0088]
二、分析样品
[0089]
(1)仪器参考条件
[0090]
a气相色谱条件
[0091]
程序升温:进样方式:不分流进样0.75min;进样量:1.0μl;进样口温度:250℃;柱流量:1.0ml/min。
[0092]
b质谱参考条件
[0093]
离子源温度:230℃;传输线温度:280℃;离子化能量:70ev;
[0094]
全扫描(scan)质量范围:50
‑
500amu;
[0095]
选择离子(sim)扫描,目标化合物扫描离子见表2。
[0096]
表2目标化合物对应的扫描离子
[0097][0098]
(2)校准
[0099]
a仪器性能检查
[0100]
仪器使用前用全氟三丁胺对质谱仪进行调谐。样品分析前以及每运12h,将1.0μl十氟三苯基膦(dftpp)使用液注入色谱,对仪器系统进行检查,所得质量离子丰度应全部符合表1中的要求。
[0101]
表1十氟三苯基膦(dftpp)关键离子及丰度标准
[0102]
质量离子丰度标准质量离子丰度标准68小于69峰的2%275基峰的10%~30%70小于69峰的2%365大于基峰的1%127基峰的40%~60%441存在且小于443峰197小于基峰的%442大于基峰的40%198基峰,丰度为100%443442峰的17%~23%
[0103]
(3)校准曲线的绘制
[0104]
分别吸取不同体积的标准和替代物标准使用液,配制成浓度为0.2、0.5、1.0、1.5、2.0μg/ml的标准系列,并同时加入内标使用液,混匀。按照仪器参考条件进行分析,得到不同目标化合物质谱图。以目标化合物浓度与内标化合物浓度的比值为横坐标,以目标化合物定量离子的响应值与内标化合物定量离子的响应值的比值为纵坐标,绘制校准曲线。
[0105]
(4)样品测定
[0106]
取待测试样按照与绘制校准曲线相同的仪器分析条件进行测定。
[0107]
(5)实验室空白试验
[0108]
在分析样品的同时,将空白试样按照与绘制校准曲线相同的仪器分析条件进行测定。
[0109]
三、结果计算与表示
[0110]
(1)定性分析
[0111]
以全扫描方式(scan)采集数据,以样品中目标化合物相对保留时间(rrt)、辅助定性离子和目标离子丰度比(q)与标准溶液中的变化范围来定性。样品中目标化合物的相对
保留时间与校准曲线该化合物的平均相对保留时间的差值应在
±
0.06内。样品中目标化合物的辅助定性离子和定量离子峰面积比(q样品)与标准曲线目标化合物的辅助定性离子和定量离子峰面积比(q标准)相对偏差控制在
±
30%以内。
[0112]
按公式(1)计算相对保留时间rrt
[0113][0114]
式中:
[0115]
rt
c
—目标化合物的保留时间,min;
[0116]
rt
is
—内标物的保留时间,min。
[0117]
平均相对保留时间(rrt):标准系列中同一目标化合物的相对保留时间平均值
[0118]
按公式(2)计算辅助定性离子和定量离子峰面积比(q)
[0119][0120]
式中:
[0121]
a
t
—定量离子峰面积;
[0122]
a
q
—辅助定性离子峰面积。
[0123]
甲基萘标准物质的选择离子扫描总离子流图,见图1。
[0124]
(2)定量分析
[0125]
以选择离子扫描方式(sim)采集数据,内标法定量。样品中目标物的质量浓度ρi(μg/l)按照公式(3)进行计算。
[0126][0127]
式中:
[0128]
ρ
i
—样品中甲基萘化合物或替代物的浓度,μg/l;
[0129]
ρ
is
—根据标准曲线查得甲基萘化合物或替代物的浓度,μg/ml;
[0130]
v—试样体积,ml;
[0131]
vt—水样体积,ml。
[0132]
最后应说明的几点是:首先,在本申请的描述中,需要说明的是,除非另有规定和限定,术语“安装”、“相连”、“连接”应做广义理解,可以是机械连接或电连接,也可以是两个元件内部的连通,可以是直接相连,“上”、“下”、“左”、“右”等仅用于表示相对位置关系,当被描述对象的绝对位置改变,则相对位置关系可能发生改变;
[0133]
以上所述仅为本发明的优选实施例而已,并不用于限制本发明,凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1