一种阿法骨化醇片有关物质的检查方法与流程
1.本发明涉及医药分析方法领域,具体涉及一种阿法骨化醇片有关物质的检查方法。
背景技术:
2.阿法骨化醇化学名为(5z,7e)-9,10-开环胆甾-5,7,10(19)-三烯-1α,3β-二醇,结构式如下:阿法骨化醇可增加小肠和肾小管对钙的重吸收,抑制甲状旁腺增生,减少甲状旁腺激素合成与释放,抑制骨吸收,增加转化生长因子-b(tgf-b)和胰岛素样生长因子-i(1gf-i)合成,促进胶原和骨基质蛋白合成,调节肌肉钙代谢,促进肌细胞分化,增强肌力,增加神经肌肉协调性,减少跌倒倾向。
3.阿法骨化醇片用于治疗改善慢性肾功能不全、甲状旁腺功能低下和抗维生素d佝偻病、骨软化症患者因维生素d代谢异常的症状,如:低钙血症、抽搐、骨痛及骨损害和骨质疏松症。
4.目前,国际上仅有《中国药典》收载了阿法骨化醇片的质量标准,但没有对有关物质的检查进行规定。而在《中国药典》中同品种软胶囊(即阿法骨化醇软胶囊)的质量标准中同样没有对有关物质的检查进行规定。对于他骨化醇类药物,《中国药典》和usp中维生素d2(麦角骨化醇)及维生素d3(胆骨化醇)的口服固体制剂标准也均未制定有关物质检查项。而针对阿法骨化醇片的有关物质检测方法及相关研究也鲜有报道。
5.化学药品研究中,有关物质的研究具有重要意义,有助于了解产品的质量及稳定性,由于阿法骨化醇片的规格仅为0.25μg和0.5μg,阿法骨化醇在片剂中占比极低,有关物质分析方法开发存在很大挑战。
6.cn 108663442 a公开了一种阿法骨化醇片有关物质的检测方法,其中的供试品的制备采用的方法为:取阿法骨化醇片50片,置于50ml纳氏比色管中,加水,涡旋至片完全崩解,加乙酸乙酯,充分振摇,静置使分层,取上层溶液减压浓缩至干,重复上述过程共三次;浓缩至干的残渣中加入一定体积的甲醇-水混合液,混匀,用玻璃注射器经0.45μm的聚四氟乙烯滤膜过滤,即得供试品溶液;精密量取100μl供试品溶液,置于10ml量瓶中,用甲醇-水混合液稀释至刻度,摇匀,作为对照溶液。但该方法需要大量的检测样品如50-100片,需要多次重复萃取操作,且检测时流动相流速为1.0~2.0ml/min,单次检测运行时间为60min,因
此溶剂量消耗比较大。现有技术cn 108663442 a中要求1%浓度水平主成分自身对照中主峰的信噪比应大于10,可见其方法定量限为1%,不能准确测定更低水平的杂质。
7.为确保药品的安全性和有效性,需要针对阿法骨化醇片开发一种专属性好、灵敏度高,准确、精密的有关物质检查的方法。本发明人经过大量的测试条件的筛选研究,惊奇的发现有通过改变供试品的制备方法,不但可以减少样品用量和溶剂消耗量,还能够大大提高检测灵敏度,克服现有技术存在的缺陷。
技术实现要素:
8.本发明的目的在于建立一种阿法骨化醇片有关物质的检查方法,该方法能够专属、灵敏、准确、精密地检测出阿法骨化醇片中的有关物质,在减少样品用量和溶剂消耗量的同事,大大提高了检测灵敏度。
9.为实现本发明的目的,提供如下实施方案。
10.在一实施方案中,本发明的一种分离测定阿法骨化醇片有关物质的方法,包括采用超高效液相色谱仪,以十八烷基硅烷键合硅胶柱为色谱柱,以乙腈、水组成流动相,采用梯度洗脱法,其特征在于:对所述片采用固相萃取技术处理制备检测样品。
11.在上述实施方案中,本发明的方法,所述采用固相萃取技术处理制备检测样品,包括以下过程,取阿法骨化醇片,研磨成粉末,称取相当于阿法骨化醇约2μg的粉末适量,加入没食子酸丙酯,再加入溶剂 1ml~2ml,使粉末呈混悬状,离心后移取上清液,再向沉淀中加入溶剂1ml~2ml,使粉末呈混悬状,离心后取上清液,与第一次离心的上清液合并,上清液用c18固相萃取小柱经淋洗和洗脱处理,去除辅料,收集洗脱液,氮气吹干,浓缩物用溶剂0.5ml复溶,离心后取上清液作为供试品,其中,所述溶剂为65-75%甲醇溶液,优选为70%甲醇溶液。
12.在上述实施方案中,本发明的方法,所述没食子酸丙酯,其加入量为4mg~10mg,所述c18固相萃取小柱经淋洗和洗脱处理,进一步可采用2根c18固相萃取小柱平行处理上清液,合并洗脱液,所述c18固相萃取小柱经淋洗和洗脱处理,淋洗液为3%~7%甲醇溶液,淋洗体积为8ml~12ml,洗脱液为乙醚,洗脱体积为7ml~9m,所述c18固相萃取小柱为waters sep-pak vac 3cc(500mg) c18 cartridges。
13.在上述实施方案中,本发明的方法,进一步包括系统适用性溶液的制备,包含:取阿法骨化醇对照品、杂质a及杂质b适量,用70%甲醇溶液溶解并稀释制成每1ml分别含阿法骨化醇、杂质a及杂质b为100μg、1μg及1μg的溶液,在80℃下水浴加热回流2小时,放冷至室温,精密量取1ml,置25ml量瓶中,用溶剂稀释至刻度,摇匀,即得系统适用性溶液,
ꢀꢀꢀꢀꢀꢀꢀ
杂质a
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
杂质b在上述实施方案中,本发明的方法,所述梯度洗脱法,选自以下两种方式之一方式一:以水为流动相a,乙腈为流动相b,梯度程序如下方式二:以76%乙腈为流动相a,95%乙腈为流动相b,梯度程序如下优选的,在上述实施方案中,本发明的方法,具体检测过程包含:精密量取系统适用性溶液和供试品溶液各20μl分别注入液相色谱仪,记录色谱图,其中,检测波长为265nm,流速为0.35ml/min~0.45ml/min,柱温为28℃~32℃。
14.在一具体实施方案中,本发明的一种阿法骨化醇片有关物质的检查方法,包含:采用反相高效液相色谱法,以十八烷基硅烷键合硅胶柱为色谱柱,优选waters uplc hss t3 2.1mm
×
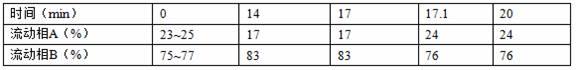
75mm,1.8μm;以乙腈和水为流动相进行梯度洗脱,所述梯度洗脱选自以下两种方式之一:方式一:以水为流动相a,乙腈为流动相b,梯度程序如下表1,表1色谱梯度洗脱方法一方式二:以76%乙腈为流动相a,95%乙腈为流动相b,梯度程序见表2,表2色谱梯度洗脱方法二其中,方式一,流动相a与流动相b起始比例为23:77~25:75,优选24:76;方式二流动相a与流动相b起始比例为100:0~94:6,优选100:0。
15.检测波长:265nm;流速:0.35ml/min~0.45ml/min,优选0.4 ml/min;进样量:20μl;
柱温:28℃~32℃,优选30℃。
16.在上述具体实施方案中,本发明的方法,进一步包括供试品溶液和系统适用性溶液的制备,包括以下过程:(1)系统适用性溶液的制备:取阿法骨化醇对照品、杂质a及杂质b适量,用溶剂(70%甲醇溶液)溶解并稀释制成每1ml分别含阿法骨化醇、杂质a及杂质b为100μg、1μg及1μg的溶液;在80℃下水浴加热回流2小时,放冷至室温,精密量取1ml,置25ml量瓶中,用溶剂稀释至刻度,摇匀,即得。
17.(2)0.25μg规格供试品溶液的制备:取阿法骨化醇片10片,置玛瑙研钵中研成粉,取粉末适量(相当于阿法骨化醇约1μg),精密称定,置10ml具塞离心管中,再加入没食子酸丙酯4mg~10mg(优选7mg)置同一玻璃离心管中,加溶剂(70%甲醇溶液)1ml~2ml,涡旋超声交替使粉末呈混悬状,离心后移取上清液,再向沉淀中加入溶剂1ml~2ml,涡旋超声交替使粉末呈混悬状,离心后取上清液,与第一次离心的上清液合并,用c18固相萃取小柱(优选waters sep-pak vac 3cc(500mg) c18 cartridges)经8ml~12ml (优选10ml)3%~7%甲醇溶液(优选5%)淋洗和7ml~9ml(优选8ml)乙醚洗脱处理,去除辅料,平行操作2份(2份合计阿法骨化醇约2μg),合并2根c18固相萃取小柱的洗脱液,氮气吹干,吹干后及时取出,精密加入溶剂0.5ml,涡旋超声交替复溶浓缩物,转移置1.5ml ep离心管中离心,取上清液作为供试品溶液。
18.或者,(3)0.5μg规格供试品溶液的制备:取阿法骨化醇片5片,置玛瑙研钵中研成粉,取粉末适量(相当于阿法骨化醇约2μg),精密称定,置10ml具塞离心管中,再加入没食子酸丙酯4mg~10mg(优选7mg)置同一玻璃离心管中,加溶剂(70%甲醇溶液)1ml~2ml,涡旋超声交替使粉末呈混悬状,离心后移取上清液,再向沉淀中加入溶剂1ml~2ml,涡旋超声交替使粉末呈混悬状,离心后取上清液,与第一次离心的上清液合并,用c18固相萃取小柱(优选waters sep-pak vac 3cc(500mg) c18 cartridges)经8ml~12ml (优选10ml)3%~7%甲醇溶液(优选5%)淋洗和7ml~9ml(优选8ml)乙醚洗脱处理,去除辅料,c18固相萃取小柱的洗脱液,氮气吹干,吹干后及时取出,精密加入溶剂0.5ml,涡旋超声交替复溶浓缩物,转移置1.5ml ep离心管中离心,取上清液作为供试品溶液。
19.在上述具体实施方案中,本发明的方法,具体检测过程包含:精密量取系统适用性溶液和供试品溶液各20μl分别注入液相色谱仪,记录色谱图。
20.检测结果:系统适用性溶液色谱图中,阿法骨化醇主峰保留时间约为10min,相对阿法骨化醇的保留时间约为0.90的峰为前阿法骨化醇峰,相对阿法骨化醇的保留时间约为0.94的峰为杂质a峰,相对阿法骨化醇的保留时间约为1.1的峰为杂质b峰;前阿法骨化醇与杂质a分离度应不小于1.3,杂质a与阿法骨化醇主峰分离度应不小于1.5,阿法骨化醇主峰与杂质b分离度应不小于1.5。供试品溶液图谱中,扣除空白辅料峰及前阿法骨化醇峰后,如有杂质峰,采用加校正因子的面积归一化法进行计算(杂质a的峰面积乘以相对响应因子0.82,其他杂质的相对响应因子为1),小于0.1%的峰可忽略不计。
21.本发明的检测方法的有益技术效果在于:(1)本发明的方法,通过排除辅料干扰及样品浓缩实现了在超低规格的阿法骨化
醇片中的有关物质检查。与现有技术cn 108663442 a相比,本发明在样品处理过程中使用的有机溶剂更少,更加绿色环保;且无需多次反复提取,因此操作性更强,能够保证方法具有稳定且较高的提取回收率,(具体验证数据参见实施例8不同提取方法的回收率、准确度对比),由于在样品处理过程中除去了大量辅料,因此也同时获得了比现有技术更干净平滑的基线、更强的专属性;此外,本发明在样品处理过程中加入了抗氧剂,能够确保样品在萃取-浓缩-复溶的过程中避免发生氧化降解,从而使检测结果能够反映样品真实的杂质水平。
22.(2)本发明的方法,采用超高效液相色谱仪进行样品分析,并采用加校正因子的面积归一化法进行计算。与现有技术cn 108663442 a相比,本发明所述的分析方法分析效率显著提高、流动相消耗大大少;且本发明采用加校正因子的面积归一化法进行杂质检出量计算,能够在确保杂质准确定量的同时,大大减少操作步骤和进样数量,成倍地提高检测效率,因此本发明经济效益更为可观。
23.(3)本品的最大日剂量是4μg,根据ich q3b指导原则,本品的报告限为0.1%,因此制剂中杂质检测方法的定量限应不低于0.1%。现有技术cn 108663442 a中要求1%浓度水平主成分自身对照中主峰的信噪比应大于10,可见其方法定量限为1%,不能很好的检测和监控0.1%~1%之间的有关物质,而本发明的方法定量限为0.1%,可以满足上述要求,具体验证数据参见实施例5 灵敏度试验。
24.(4)本发明的方法中的色谱部分,因具有超高效液相色谱多快好省的特点,可替代各药典中收载的阿法骨化醇原料药有关物质分析方法,由此可以得到和片剂有关物质检查具有可比性的杂质谱。
25.综上,本发明建立的阿法骨化醇片有关物质的检测方法,按国内外相关指导原则进行了方法验证,表明本方法专属性强,精密度好,准确度且灵敏度高,且在整体设计上使方法具有效率高、操作性强、安全环保的优点,可用于阿法骨化醇片的质量研究,并对类似品种具有借鉴价值。
附图说明
26.图1为系统适用性溶液色谱图,图2为供试品溶液色谱图,图3为实施例4的阿法骨化醇线性结果图,图4为实施例4的杂质a的线性结果图,图5为实施例4的杂质b的线性结果图。
具体实施方式
27.下面通过实施例对本发明做进一步的说明,以帮助理解本发明的实质,但不以任何方式限制本发明的范围。
28.实施例1 色谱条件的耐用性试验本试验选用waters uplc hss t3 2.1mm
×
75mm,1.8μm色谱柱,检测波长为265nm,以水为流动相a,乙腈为流动相b,通过调整流动相比例、流速、柱温来筛选本试验的色谱方法。具体方案见表3。
29.表3 色谱条件耐用性考察方案溶液的配制:(1)系统适用性溶液的制备:取阿法骨化醇对照品、杂质a及杂质b适量,用溶剂(70%甲醇溶液)溶解并稀释制成每1ml分别含阿法骨化醇、杂质a及杂质b为100μg、1μg及1μg的溶液;在80℃下水浴加热回流2小时,放冷至室温,精密量取1ml,置25ml量瓶中,用溶剂稀释至刻度,摇匀,即得。
30.(2)0.25μg规格供试品溶液的制备:取阿法骨化醇片10片,置玛瑙研钵中研成粉,取粉末适量(相当于阿法骨化醇约1μg),精密称定,置10ml具塞离心管中,再加入没食子酸丙酯7mg置同一玻璃离心管中,加溶剂(70%甲醇溶液)1ml~2ml,涡旋超声交替使粉末呈混悬状,离心后移取上清液,再向沉淀中加入溶剂1ml~2ml,涡旋超声交替使粉末呈混悬状,离心后取上清液,与第一次离心的上清液合并,用c18固相萃取小柱(waters sep-pak vac 3cc(500mg) c18 cartridges)经10ml 5%甲醇溶液淋洗和8ml乙醚洗脱处理,去除辅料,平行操作2份(2份合计阿法骨化醇约2μg),合并2根c18固相萃取小柱的洗脱液,氮气吹干,吹干后及时取出,精密加入溶剂(70%甲醇溶液)0.5ml,涡旋超声交替复溶浓缩物,转移置1.5ml ep离心管中离心,取上清液作为供试品溶液。
31.样品测定:精密量取系统适用性溶液、供试品溶液各20μl,按相应色谱条件注入液相色谱仪中,记录色谱图。考察各色谱条件下1)系统适用性溶液色谱图中前阿法骨化醇与杂质a的分离度、杂质a与阿法骨化醇主峰的分离度及阿法骨化醇主峰与杂质b的分离度;2)出峰顺序是否改变;3)供试品溶液色谱图中,各杂质含量与方案1的杂质含量之比。结果见表4、表5。
32.表4 色谱条件耐用性考察结果—系统适用性溶液表5 色谱条件耐用性考察结果—供试品溶液
结果表明,改变各色谱条件,系统适用性溶液中前阿法骨化醇与杂质a分离度均为1.4,不小于1.3,杂质a与阿法骨化醇主峰分离度为2.0~2.1,不小于1.5,阿法骨化醇主峰与杂质b分离度为3.1~3.2,不小于1.5;各杂质出峰顺序未改变;供试品溶液中仅存的一个未知杂质的含量与方案1测定结果之比在97.4%~105.3%之间。本方法色谱条件具有较好耐用性。
33.实施例2 固相萃取条件的耐用性色谱条件:同实施例1的方案1。
34.溶液的配制:(1)系统适用性溶液的制备:同实施例1。
35.(2)0.25μg规格供试品溶液的制备:取阿法骨化醇片10片,置玛瑙研钵中研成粉,取粉末适量(相当于阿法骨化醇约1μg),精密称定,置10ml具塞离心管中,再加入没食子酸丙酯7mg置同一玻璃离心管中,加溶剂(70%甲醇溶液)1ml,涡旋超声交替使粉末呈混悬状,离心后移取上清液,再向沉淀中加入溶剂1ml,涡旋超声交替使粉末呈混悬状,离心后取上清液,与第一次离心的上清液合并,用c18固相萃取小柱(waters sep-pak vac 3cc(500mg) c18 cartridges)经5%甲醇溶液淋洗和乙醚洗脱处理,去除辅料,平行操作2份(2份合计阿法骨化醇约2μg),合并2根c18固相萃取小柱的洗脱液,氮气吹干,吹干后及时取出,精密加入溶剂(70%甲醇溶液)0.5ml,涡旋超声交替复溶浓缩物,转移置1.5ml ep离心管中离心,取上清液作为供试品溶液。
36.通过改变淋洗体积和洗脱体积来筛选本试验的色谱方法。具体方案见表6。
37.表6 固相萃取条件耐用性考察方案样品测定:精密量取系统适用性溶液、各供试品溶液各20μl,注入液相色谱仪中,记录色谱图。考察各固相萃取条件的供试品溶液色谱图中,各杂质含量与方案1的杂质含量之比。结果见表7。
38.表7 固相萃取条件耐用性考察结果
结果表明,改变固相萃取条件,即改变淋洗体积和洗脱体积,供试品溶液中仅存的一个未知杂质的含量与正常条件测定结果的比为97.4%~105.3%。固相萃取条件具有较好耐用性。
39.实施例3 供试品溶液的稳定性色谱条件:同实施例1方案1。
40.溶液的配制:同实施例1样品测定:取在室温下避光放置的供试品溶液,分别于0h、2h、4h、8h、12h、16h、20h及24h精密量取20μl注入液相色谱仪中,记录色谱图。考察放置不同时间的供试品溶液色谱图中,各杂质含量与0h杂质含量之比。结果见表8。
41.表8 供试品溶液稳定性考察结果结果显示,供试品溶液在24h内稳定。
42.实施例4 标准曲线的及已知杂质的校正因子色谱条件:同实施例1方案1。
43.溶液的配制:(1)杂质a线性储备液:取杂质a对照品2.083mg,精密称定,置20ml棕色量瓶中,用乙醇溶解并稀释至刻度,摇匀,精密移取1ml,置50ml棕色量瓶中,用溶剂(70%甲醇溶液)稀释至刻度,摇匀,即得。(含杂质a 2.079μg/ml)(2)杂质b线性储备液:取杂质b对照品2.036mg,精密称定,置20ml棕色量瓶中,用乙醇溶解并稀释至刻度,摇匀精密移取1ml,置50ml棕色量瓶中,用溶剂稀释至刻度,摇匀,即得。(含杂质b 1.964μg/ml)(3)阿法骨化醇线性储备液-1:取阿法骨化醇工作对照品1.992mg,精密称定,置20ml棕色量瓶中,用乙醇溶解并稀释至刻度,摇匀,即得。(含阿法骨化醇98.31μg/ml)(4)阿法骨化醇线性储备液-2:精密移取阿法骨化醇线性储备液-1 1ml置50ml棕色量瓶中,用溶剂(70%甲醇溶液)稀释至刻度,摇匀,即得。(含阿法骨化醇1.966μg/ml)
(5)混合溶液:分别精密移取杂质a线性储备液、杂质b线性储备液和阿法骨化醇线性储备液-2 5ml,置50ml棕色量瓶中,用溶剂(70%甲醇溶液)稀释至刻度,摇匀,即得。(含阿法骨化醇0.1966μg/ml、含杂质a 0.2079μg/ml、含杂质b 0.1964μg/m)取上述混合溶液,照下表用溶剂稀释制成系列浓度的线性供试品溶液:表9 线性供试品溶液配制阿法骨化醇扩大线性范围至150%浓度水平,详细配制过程如下:(6)50%浓度阿法骨化醇线性溶液:同阿法骨化醇线性储备液-2。(含阿法骨化醇1.966μg/ml)(7)100%浓度阿法骨化醇线性溶液:精密量取阿法骨化醇线性储备液-1 1ml置25ml棕色量瓶中,用70%甲醇稀释至刻度,摇匀,即得。(含阿法骨化醇3.932μg/ml)(8)150%浓度阿法骨化醇线性溶液:精密量取阿法骨化醇线性储备液-1 3ml置50ml棕色量瓶中,用70%甲醇稀释至刻度,摇匀,即得。(含阿法骨化醇5.898μg/ml)样品测定:精密量取各线性供试品溶液20μl,注入液相色谱仪中,记录色谱图。考察各已知杂质及阿法骨化醇峰面积与浓度线性关系及已知杂质的相对响应因子。结果见表10。
44.表10 标准曲线的及已知杂质的校正因子考察结果
表10结果表明:(1)阿法骨化醇在0.0039μg/ml(0.1%)~5.898μg/ml(150%)范围内,线性方程为y = 116,414.3242x + 123.7802,线性相关系数为1.0000,峰面积与浓度线性关系良好;(2)杂质a在0.0042μg/ml(0.1%)~0.0832μg/ml(2%)范围内,线性方程为y = 141,306.3706x + 15.5622,线性相关系数为1.0000,峰面积与浓度线性关系良好,杂质a校正因子为0.82;
(3)杂质b在0.0039μg/ml(0.1%)~0.0785μg/ml(2%)范围内,线性方程为y = 115,129.9449x
ꢀ‑ꢀ
74.6833,线性相关系数为1.0000,峰面积与浓度线性关系良好,杂质b校正因子为1.0。
45.实施例5 灵敏度试验色谱条件:同实施例1方案1。
46.溶液配制:(1)定量限溶液:同实施例4表9中1号线性溶液。
47.(2)检测限溶液:精密量取定量限溶液5ml,置10ml量瓶中,用溶剂(70%甲醇溶液)稀释至刻度,摇匀,即得。
48.样品测定:(1)精密量取定量限溶液20μl,注入液相色谱仪中,记录色谱图,连续进样6针。考察阿法骨化醇峰、杂质a峰、杂质b峰的信噪比(s/n)及峰面积的rsd。
49.(2)精密量取检测限溶液20μl,注入液相色谱仪中,记录色谱图。考察阿法骨化醇峰、杂质a峰、杂质b峰的信噪比(s/n)。结果见表11。
50.表11 灵敏度试验考察结果结果表明,定量限溶液连续进样6针,阿法骨化醇、杂质a、杂质b的s/n均大于10,峰面积的rsd分别为6.9%、5.1%、7.6%;检测限溶液进样1针,阿法骨化醇、杂质a、杂质b的s/n不小于3。本方法具有良好的检测灵敏度。
51.实施例6 重复性试验色谱条件:同实施例1方案1。
52.溶液配制:(1)系统适用性溶液的制备:同实施例1。
53.(2)0.5μg规格供试品溶液的制备:取阿法骨化醇片5片,置玛瑙研钵中研成粉,取粉末适量(相当于阿法骨化醇约2μg),精密称定,置10ml具塞离心管中,再加入没食子酸丙酯4mg~10mg(优选7mg)置同一玻璃离心管中,加溶剂(70%甲醇溶液)1ml~2ml,涡旋超声交替使粉末呈混悬状,离心后移取上清液,再向沉淀中加入溶剂1ml~2ml,涡旋超声交替使粉末呈混悬状,离心后取上清液,与第一次离心的上清液合并,用c18固相萃取小柱(优选waters sep-pak vac 3cc(500mg) c18 cartridges)经10ml 5%甲醇溶液淋洗和8ml乙醚洗脱处理,去除辅料,c18固相萃取小柱的洗脱液,氮气吹干,吹干后及时取出,精密加入溶剂(70%甲醇
溶液)0.5ml,涡旋超声交替复溶浓缩物,转移置1.5ml ep离心管中离心,取上清液作为供试品溶液。平行配制6份。
54.样品测定:精密量取系统适用性溶液、供试品溶液各20μl,注入液相色谱仪中,记录色谱图。考察样品配制的精密度。结果见表12。
55.表12 重复性试验考察结果结果表明,供试品溶液中仅存的一个未知杂质检出量的rsd为4.4%。本方法具有良好的重复性。
56.实施例7 样品的测定色谱条件:同实施例1中方案1。
57.溶液的配制:(1)系统适用性溶液的制备:同实施例1。
58.(2)0.25μg规格供试品溶液的制备:同实施例1。
59.(3)0.5μg规格供试品溶液的制备:同实施例6。
60.样品测定:精密量取系统适用性溶液、供试品溶液各20μl,注入液相色谱仪中,记录色谱图。结果见表13。
61.表13 样品测定考察结果实施例8 不同提取方法的回收率(准确度)对比方法一、固相萃取法:溶液的配制:(1)阿法骨化醇储备液:取阿法骨化醇对照品2mg,精密称定,置100ml棕色量瓶中,用乙醇溶解并稀释至刻度,摇匀,即得。
62.(2)杂质a储备液:取杂质a对照品2mg,精密称定,置20ml棕色量瓶中,用乙醇溶解并稀释至刻度,摇匀;精密移取1ml,置50ml棕色量瓶中,用70%甲醇稀释至刻度,摇匀,即得。
63.(3)杂质b储备液:取杂质b对照品2mg,精密称定,置20ml棕色量瓶中,用乙醇溶解并稀释至刻度,摇匀;精密移取1ml,置50ml棕色量瓶中,用70%甲醇稀释至刻度,摇匀,即得。
64.(4)杂质a、b混合溶液:分别精密移取杂质a储备液和杂质b储备液10ml,置100ml棕色量瓶中,用70%甲醇稀释至刻度,摇匀,即得。
65.(5)0.5%浓度水平溶液:精密量取杂质a、b混合溶液0.5ml,置20ml棕色量瓶中,再精密移取阿法骨化醇储备液1ml,置同一量瓶中,用70%甲醇稀释至刻度,摇匀,即得。
66.(6)1.0%浓度水平溶液:精密量取杂质a、b混合溶液1ml,置20ml棕色量瓶中,再精密移取阿法骨化醇储备液1ml,置同一量瓶中,用70%甲醇稀释至刻度,摇匀,即得。
67.(7)1.5%浓度水平溶液:分别精密量取杂质a、b储备液各1.5ml,置20ml棕色量瓶中,再精密移取阿法骨化醇储备液1ml,置同一量瓶中,用70%甲醇稀释至刻度,摇匀,即得。
68.(8)0.5%浓度水平准确度溶液:取空白辅料约340mg,精密称定,置10ml具塞玻璃离心管中,再加入没食子酸丙酯6.8mg,加入0.5%浓度水平溶液1ml,后续操作同实施例1。(平行配制3份)(9)1.0%浓度水平准确度溶液:取空白辅料约340mg,精密称定,置10ml具塞玻璃离心管中,再加入没食子酸丙酯约6.8mg,加入1.0%浓度水平溶液1ml,后续操作同实施例1。(平行配制3份)(10)1.5%浓度水平准确度溶液:取空白辅料约340mg,精密称定,置10ml玻璃离心管中,再加入没食子酸丙酯6.8mg约,加入1.5%浓度水平溶液1ml,后续操作同实施例1。(平行配制3份)样品测定:精密量取准确度供试品溶液各20μl,注入液相色谱仪中,记录色谱图。按加校正因子的面积归一化法计算3个浓度水平共9份准确度溶液中已知杂质各自的回收率,以及9份供试品溶液已知杂质的平均回收率及回收率的rsd。结果见表14~15。
69.表14杂质a准确度结果表15杂质b准确度结果
结果表明,9份供试品溶液杂质a平均回收率为83.1%,rsd为3.2%;杂质b平均回收率为94.5%,rsd为3.4%。方法准确度良好。
70.方法二、有机溶剂萃取法:溶液的配制:(1)阿法骨化醇对照品溶液:取阿法骨化醇对照品2mg,精密称定,置20ml棕色量瓶中,用乙醇溶解并稀释至刻度,摇匀;精密量取1ml,置20ml棕色量瓶中,用70%甲醇稀释至刻度,摇匀,即得。
71.(2)准确度储备液:取阿法骨化醇对照品2mg,精密称定,置20ml棕色量瓶中,用乙醇溶解并稀释至刻度,摇匀;精密量取1ml,置20ml棕色量瓶中,用乙酸乙酯稀释至刻度,摇匀,即得。
72.(3)准确度溶液:取空白片剂研磨,取10片量置50ml棕色量瓶中,精密加入准确度储备液1ml与空白辅料混合后氮气吹干。加水10ml,涡旋1min,加乙酸乙酯10ml,涡旋1min,静置后将乙酸乙酯层转移至玻璃试管,剩余残渣再次用乙酸乙酯提取2次,合并乙酸乙酯层,氮气吹干,精密加入70%甲醇1ml,涡旋超声交替复溶浓缩物,转移置1.5ml ep离心管中离心,取上清液作为准确度溶液。(平行配制2份)样品测定:精密量取对照品溶液、准确度溶液各20μl,注入液相色谱仪中,记录色谱图。考察主成分的回收率。结果见表16。
73.表16有机溶剂萃取法回收率考察实验结果结果表明,有机溶剂萃取法回收率较低,且平行性不佳。采用有机溶剂萃取法处理样品时,为解除辅料的包裹,需要先用水将片剂中大量的水溶性辅料溶解,再用有机溶剂进行液液萃取,但由于主成分在水中具有一定的溶解度且剂量很低,且可能存在辅料与主成分在有机相中的竞争溶解,因此无法使用有机溶剂将主成分及其有关物质完全提取出来,而其较低且平行性不佳的回收率无法保证制剂中有关物质的准确检出,因此不适用于本品有关物质的检测。
74.以上所述的实施例仅仅是对本发明的优选实施方式进行描述,并非对本发明的范围进行限定,在不脱离本发明设计精神的前提下,本领域普通技术人员对本发明的技术方案做出的各种变形和改进,均应落入本发明权利要求书确定的保护范围内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1