一种茚虫威关键中间体含量的分析方法与流程
1.本发明涉及化工分析技术领域,具体涉及一种茚虫威关键中间体含量的分析方法。
背景技术:
2.茚虫威是美国杜邦公司研发的一种新型、高效、低毒噁二嗪类杀虫剂,具有触杀与胃毒双重功效,能够有效解决抗性害虫。与其他杀虫剂如菊酯、有机磷、氨基甲酸酯类均无交互抗性,可以很好地解决目前市场上难防的稻纵卷叶螟、二化螟及抗性小菜蛾问题。此外,茚虫威具有极宽的杀虫谱,一药多防,在防治夜蛾类害虫的同时,对盲蝽蟓等还具有很好的抑制作用,因此是一种很好的综合治理工具,可以很好地解决目前多种农药混用后的残留及环境污染问题。茚虫威由于其独特的作用机制,因此市场前景广阔。
[0003]7‑
氯茚并[1,2
‑
e][1,3,4]恶二嗪
‑
2,4a(3h,5h)
‑
二羧酸
‑
4a
‑
甲酯
‑2‑
苄酯是茚虫威的中间体,目前准确检测7
‑
氯茚并[1,2
‑
e][1,3,4]恶二嗪
‑
2,4a(3h,5h)
‑
二羧酸
‑
4a
‑
甲酯
‑2‑
苄酯含量的分析方法还处在空白状态,并且它的含量准确性直接影响其下游产品的准确度,进而影响茚虫威含量的准确性。寻找一个稳定准确检测7
‑
氯茚并[1,2
‑
e][1,3,4]恶二嗪
‑
2,4a(3h,5h)
‑
二羧酸
‑
4a
‑
甲酯
‑2‑
苄酯含量的分析方法对于保证茚虫威产品质量具有重要作用和现实意义。
技术实现要素:
[0004]
针对现有技术中存在的问题,本发明提供一种茚虫威关键中间体含量的分析方法,填补了技术空白,专属性强,精密度好,回收率高,可信度高,重复性好,特别适用于农药原药产品的质量控制。
[0005]
本发明的技术方案为:
[0006]
一种茚虫威关键中间体含量的分析方法,即采用高效液相色谱分析方法对茚虫威关键中间体7
‑
氯茚并[1,2
‑
e][1,3,4]恶二嗪
‑
2,4a(3h,5h)
‑
二羧酸
‑
4a
‑
甲酯
‑2‑
苄酯的含量进行分析测定,具体方法如下:
[0007]
(1)用甲醇作溶剂分别溶解标准品和待测样品,配制标准品溶液和待测样品溶液,标准品溶液和待测样品溶液的浓度范围均为0.5
‑
1mg/ml;
[0008]
(2)将高效液相色谱的检测波长设定至230nm
‑
300nm内,仪器基线稳定后按标准品、待测样品、待测样品、标准品的顺序依次进样,分别计算标准品溶液、待测样品溶液的7
‑
氯茚并[1,2
‑
e][1,3,4]恶二嗪
‑
2,4a(3h,5h)
‑
二羧酸
‑
4a
‑
甲酯
‑2‑
苄酯峰面积平均值;
[0009]
(3)按照外标法公式对待测样品溶液中7
‑
氯茚并[1,2
‑
e][1,3,4]恶二嗪
‑
2,4a(3h,5h)
‑
二羧酸4a
‑
甲酯
‑2‑
苄酯的质量分数x1进行计算,具体公式如下:
[0010][0011]
式中,
[0012]
a1——标准品溶液中7
‑
氯茚并[1,2
‑
e][1,3,4]恶二嗪
‑
2,4a(3h,5h)
‑
二羧酸
‑
4a
‑
甲酯
‑2‑
苄酯峰面积的平均值,
[0013]
a2——待测样品溶液中7
‑
氯茚并[1,2
‑
e][1,3,4]恶二嗪
‑
2,4a(3h,5h)
‑
二羧酸
‑
4a
‑
甲酯
‑2‑
苄酯峰面积的平均值,
[0014]
m1——标准品的质量,
[0015]
m2——待测样品的质量,
[0016]
p1——标准品中7
‑
氯茚并[1,2
‑
e][1,3,4]恶二嗪
‑
2,4a(3h,5h)
‑
二羧酸
‑
4a
‑
甲酯
‑2‑
苄酯的质量分数;
[0017]
其中,高效液相色谱条件包括:
[0018]
色谱柱为ods
‑
c18反相色谱柱,色谱柱温度为30℃
‑
50℃,流动相为甲醇和冰乙酸水溶液的混合体系。本发明人经过大量的实验,发现在流动相的选择上,选用甲醇与冰乙酸水溶液可以很好地将目标物与杂质有效的分离,并且在经济适用性上优于其他溶剂。
[0019]
优选的,高效液相色谱的检测波长为280nm。280nm检测波长对7
‑
氯茚并[1,2
‑
e][1,3,4]恶二嗪
‑
2,4a(3h,5h)
‑
二羧酸
‑
4a
‑
甲酯2
‑
苄酯来说是最稳定的紫外吸收波长,在此波长下测定误差小。
[0020]
优选的,色谱柱的柱长为150mm,柱内径为4.5
‑
5mm,更优选4.6mm,柱粒度为3.5μm。
[0021]
优选的,色谱柱的温度为40℃。
[0022]
优选的,所述混合体系中冰乙酸水溶液的体积分数为20
‑
50%,更优选为25%。
[0023]
优选的,所述高效液相色谱条件还包括:每次进样的样品体积为5μl,流动相的流速为1ml/min。
[0024]
本发明的有益效果在于,
[0025]
(1)本发明采用高效液相色谱对7
‑
氯茚并[1,2
‑
e][1,3,4]恶二嗪
‑
2,4a(3h,5h)
‑
二羧酸
‑
4a
‑
甲酯
‑2‑
苄酯的含量进行检测,包括色谱柱为ods
‑
c18反相色谱柱,色谱柱温度为30℃
‑
50℃,流动相为甲醇和冰乙酸水溶液的混合体系,检测波长为230nm
‑
300nm,能够准确测定其含量,填补了现有技术中相应领域的技术空白;
[0026]
(2)采用上述方法检测7
‑
氯茚并[1,2
‑
e][1,3,4]恶二嗪
‑
2,4a(3h,5h)
‑
二羧酸
‑
4a
‑
甲酯
‑2‑
苄酯的质量分数稳定准确,提高了检测的精度和稳定性,可以实现杂质、主峰分离完全,色谱峰形好,保留时间稳定,积分计算结果准确、重复性好,所得结果可信度高,特别适用于农药原药中间产品的质量控制,对保证最终产品的质量具有重要作用和现实意义;同时所得结果更加准确及时,为茚虫威的生产提供了有力的数据支持。
附图说明
[0027]
为了更清楚地说明本发明实施例或现有技术中的技术方案,下面将对实施例或现有技术描述中所需要使用的附图作简单地介绍,显而易见地,对于本领域普通技术人员而言,在不付出创造性劳动的前提下,还可以根据这些附图获得其他的附图。
[0028]
图1是实施例1中标准品溶液的色谱图;
[0029]
图2是实施例1中待测样品溶液的色谱图;
[0030]
图3是实施例2中标准品溶液的色谱图;
[0031]
图4是实施例2中待测样品溶液的色谱图;
[0032]
图5是实施例3中标准品溶液的色谱图;
[0033]
图6是实施例3中待测样品溶液的色谱图;
[0034]
图7是试验例4中的线性关系图;
[0035]
图8是对比例1中波长是260nm线性关系图;
[0036]
图9是对比例1中波长是300nm线性关系图;
[0037]
图10是对比例2中流动相比例为甲醇:0.8%乙酸水溶液=90:10时的色谱图;
[0038]
图11是对比例2中流动相比例为甲醇:0.8%乙酸水溶液=60:40时的色谱图;
[0039]
其中,
[0040]
图1
‑
6的色谱图中,横坐标代表时间,纵坐标代表吸光度;
[0041]
图8
‑
9的线性关系图中,横坐标代表标准品溶液浓度(单位为g/l),纵坐标代表峰面积;
[0042]
图10
‑
11的色谱图中,横坐标代表时间,纵坐标代表吸光度。
具体实施方式
[0043]
为了使本技术领域的人员更好地理解本发明中的技术方案,下面将结合本发明实施例中的附图,对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都应当属于本发明保护的范围。
[0044]
实施例1
[0045]
生产中210批,得到产品1960公斤7
‑
氯茚并[1,2
‑
e][1,3,4]恶二嗪
‑
2,4a(3h,5h)
‑
二羧酸
‑
4a
‑
甲酯2
‑
苄酯的固体样品,对产品中7
‑
氯茚并[1,2
‑
e][1,3,4]恶二嗪
‑
2,4a(3h,5h)
‑
二羧酸
‑
4a
‑
甲酯2
‑
苄酯含量进行分析,方法包括如下步骤:
[0046]
(1)准确称取7
‑
氯茚并[1,2
‑
e][1,3,4]恶二嗪
‑
2,4a(3h,5h)
‑
二羧酸
‑
4a
‑
甲酯2
‑
苄酯标准品置于100ml容量瓶中,加90ml甲醇,振荡溶解后,用甲醇稀释至刻度,得到标准品溶液备用,标准品中7
‑
氯茚并[1,2
‑
e][1,3,4]恶二嗪
‑
2,4a(3h,5h)
‑
二羧酸
‑
4a
‑
甲酯2
‑
苄酯的质量分数p1=98.6%;
[0047]
准确称取待测样品置于100ml容量瓶中,加90ml甲醇,振荡溶解后,用甲醇稀释至刻度,得到待测样品溶液备用;
[0048]
(2)采用高效液相色谱仪,色谱柱为ods
‑
c18色谱柱,色谱柱的柱长为150mm,柱内径为4.6mm,柱粒度为3.5μm;色谱柱的温度为40℃;流动相为甲醇和0.8%冰乙酸水溶液的混合体系,混合体系中冰乙酸水溶液的体积分数为25%;每次进样的样品体积为5μl,流动相的流速为1ml/min;检测波长设定为280nm;
[0049]
(3)仪器基线稳定后按标准品、待测样品、待测样品、标准品的顺序依次进样,分别计算标准品溶液、待测样品溶液的7
‑
氯茚并[1,2
‑
e][1,3,4]恶二嗪
‑
2,4a(3h,5h)
‑
二羧酸
‑
4a
‑
甲酯2
‑
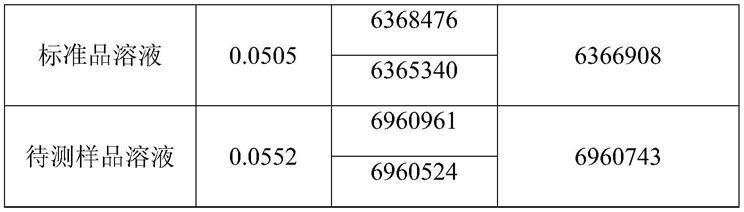
苄酯峰面积平均值,检测数据如下表1所示:
[0050]
表1实施例1检测结果
[0051]
[0052][0053]
(4)按照外标法公式对待测样品中7
‑
氯茚并[1,2
‑
e][1,3,4]恶二嗪
‑
2,4a(3h,5h)
‑
二羧酸
‑
4a
‑
甲酯
‑2‑
苄酯的质量分数进行计算,计算得到待测样品中7
‑
氯茚并[1,2
‑
e][1,3,4]恶二嗪
‑
2,4a(3h,5h)
‑
二羧酸
‑
4a
‑
甲酯2
‑
苄酯的质量分数为98.6%。
[0054]
具体公式如下,
[0055][0056]
式中,
[0057]
a1——标准品溶液中7
‑
氯茚并[1,2
‑
e][1,3,4]恶二嗪
‑
2,4a(3h,5h)
‑
二羧酸
‑
4a
‑
甲酯
‑2‑
苄酯峰面积的平均值,
[0058]
a2——待测样品溶液中7
‑
氯茚并[1,2
‑
e][1,3,4]恶二嗪
‑
2,4a(3h,5h)
‑
二羧酸
‑
4a
‑
甲酯
‑2‑
苄酯峰面积的平均值,
[0059]
m1——标准品的质量,
[0060]
m2——待测样品的质量,
[0061]
p1——标准品中7
‑
氯茚并[1,2
‑
e][1,3,4]恶二嗪
‑
2,4a(3h,5h)
‑
二羧酸
‑
4a
‑
甲酯
‑2‑
苄酯的质量分数,
[0062]
x1——待测样品中7
‑
氯茚并[1,2
‑
e][1,3,4]恶二嗪
‑
2,4a(3h,5h)
‑
二羧酸
‑
4a
‑
甲酯
‑2‑
苄酯的质量分数;
[0063]
图1为本实施例中标准品溶液的色谱图,图中7.923min位置对应的峰为7
‑
氯茚并[1,2
‑
e][1,3,4]恶二嗪
‑
2,4a(3h,5h)
‑
二羧酸
‑
4a
‑
甲酯
‑2‑
苄酯特征峰;图2为本实施例中测定的待测样品溶液的色谱图,7.924min位置对应的峰为7
‑
氯茚并[1,2
‑
e][1,3,4]恶二嗪
‑
2,4a(3h,5h)
‑
二羧酸
‑
4a
‑
甲酯2
‑
苄酯。
[0064]
实施例2
[0065]
小试样品20201208批,得到固体98.4g,对产品中7
‑
氯茚并[1,2
‑
e][1,3,4]恶二嗪
‑
2,4a(3h,5h)
‑
二羧酸
‑
4a
‑
甲酯2
‑
苄酯含量进行分析,方法包括如下步骤:
[0066]
(1)准确称取7
‑
氯茚并[1,2
‑
e][1,3,4]恶二嗪
‑
2,4a(3h,5h)
‑
二羧酸
‑
4a
‑
甲酯2
‑
苄酯标准品置于100ml容量瓶中,加90ml甲醇,振荡溶解后,用甲醇稀释至刻度,得到标准品溶液备用,标准品中7
‑
氯茚并[1,2
‑
e][1,3,4]恶二嗪
‑
2,4a(3h,5h)
‑
二羧酸
‑
4a
‑
甲酯2
‑
苄酯的质量分数p1=98.6%;
[0067]
准确称取待测样品置于100ml容量瓶中,加90ml甲醇,振荡溶解后,用甲醇稀释至刻度,得到待测样品溶液备用;
[0068]
(2)采用高效液相色谱仪,色谱柱为ods
‑
c18反相色谱柱,色谱柱的柱长为150mm,柱内径为4.6mm,柱粒度为3.5μm;色谱柱的温度为40℃;流动相为甲醇和0.8%冰乙酸水溶液的混合体系,混合体系中冰乙酸水溶液的体积分数为25%;每次进样的样品体积为5μl,
流动相的流速为1ml/min;检测波长设定为280nm;
[0069]
(3)仪器基线稳定后按标准品、待测样品、待测样品、标准品的顺序依次进样,分别计算标准品溶液、待测样品溶液的7
‑
氯茚并[1,2
‑
e][1,3,4]恶二嗪
‑
2,4a(3h,5h)
‑
二羧酸
‑
4a
‑
甲酯2
‑
苄酯峰面积平均值,检测数据如下表2所示:
[0070]
表2实施例2检测结果
[0071][0072][0073]
(4)按照外标法公式对待测样品溶液中7
‑
氯茚并[1,2
‑
e][1,3,4]恶二嗪
‑
2,4a(3h,5h)
‑
二羧酸
‑
4a
‑
甲酯
‑2‑
苄酯的质量分数进行计算,具体公式同实施例1,计算得到待测样品溶液中7
‑
氯茚并[1,2
‑
e][1,3,4]恶二嗪
‑
2,4a(3h,5h)
‑
二羧酸
‑
4a
‑
甲酯
‑2‑
苄酯的质量分数为98.8%。
[0074]
图3是本实施例中标准品溶液的色谱图,图中7.368min位置对应的峰为7
‑
氯茚并[1,2
‑
e][1,3,4]恶二嗪
‑
2,4a(3h,5h)
‑
二羧酸
‑
4a
‑
甲酯
‑2‑
苄酯,图4是本实施例中待测样品溶液的色谱图,图中7.342min位置对应的峰为7
‑
氯茚并[1,2
‑
e][1,3,4]恶二嗪
‑
2,4a(3h,5h)
‑
二羧酸
‑
4a
‑
甲酯2
‑
苄酯。
[0075]
实施例3
[0076]
生产中203批,得到产品1820公斤7
‑
氯茚并[1,2
‑
e][1,3,4]恶二嗪
‑
2,4a(3h,5h)
‑
二羧酸
‑
4a
‑
甲酯
‑2‑
苄酯的固体样品,对产品中7
‑
氯茚并[1,2
‑
e][1,3,4]恶二嗪
‑
2,4a(3h,5h)
‑
二羧酸
‑
4a
‑
甲酯
‑2‑
苄酯含量进行分析,方法包括如下步骤:
[0077]
(1)准确称取7
‑
氯茚并[1,2
‑
e][1,3,4]恶二嗪
‑
2,4a(3h,5h)
‑
二羧酸
‑
4a
‑
甲酯
‑2‑
苄酯标准品置于100ml容量瓶中,加90ml甲醇,振荡溶解后,用甲醇稀释至刻度,得到标准品溶液备用,标准品中7
‑
氯茚并[1,2
‑
e][1,3,4]恶二嗪
‑
2,4a(3h,5h)
‑
二羧酸
‑
4a
‑
甲酯2
‑
苄酯的质量分数p1=98.6%;
[0078]
准确称取待测样品置于100ml容量瓶中,加100ml甲醇,振荡溶解后,用甲醇稀释至刻度,得到待测样品溶液备用;
[0079]
(2)采用高效液相色谱仪带二极管阵列检测器,色谱柱为ods
‑
c18手性色谱柱,色谱柱的柱长为150mm,柱内径为4.6mm,柱粒度为3.5μm;色谱柱的温度为40℃;流动相为甲醇和0.8%冰乙酸水溶液的混合体系,混合体系中冰乙酸水溶液的体积分数为25%;每次进样的样品体积为5μl,流动相的流速为1ml/min;检测波长设定为280nm;
[0080]
(3)仪器基线稳定后按标准品、待测样品、标准品、待测样品的顺序依次进样,分别
计算标准品溶液、待测样品溶液的7
‑
氯茚并[1,2
‑
e][1,3,4]恶二嗪
‑
2,4a(3h,5h)
‑
二羧酸
‑
4a
‑
甲酯2
‑
苄酯峰面积平均值,检测数据如下表3所示:
[0081]
表3实施例3检测结果
[0082][0083]
(4)按照外标法公式对待测样品中7
‑
氯茚并[1,2
‑
e][1,3,4]恶二嗪
‑
2,4a(3h,5h)
‑
二羧酸
‑
4a
‑
甲酯
‑2‑
苄酯的质量分数进行计算,具体公式同实施例1,计算得到待测样品中7
‑
氯茚并[1,2
‑
e][1,3,4]恶二嗪
‑
2,4a(3h,5h)
‑
二羧酸
‑
4a
‑
甲酯
‑2‑
苄酯的质量分数为98.4%。
[0084]
图5是本实施例中标准品溶液的色谱图,图中7.263min位置对应的峰为7
‑
氯茚并[1,2
‑
e][1,3,4]恶二嗪
‑
2,4a(3h,5h)
‑
二羧酸
‑
4a
‑
甲酯2
‑
苄酯,图6是本实施例中待测样品溶液的色谱图,图中7.267min位置对应的峰为7
‑
氯茚并[1,2
‑
e][1,3,4]恶二嗪
‑
2,4a(3h,5h)
‑
二羧酸
‑
4a
‑
甲酯
‑2‑
苄酯。
[0085]
试验例1稳定性试验
[0086]
取实施例1中标准品溶液为考察对象,室温下每间隔一定的时间分析一次,共分析5次,并同时记录峰面积,分析条件包括:采用高效液相色谱仪带二极管阵列检测器,色谱柱为ods
‑
c18反相色谱柱,色谱柱的柱长为150mm,柱内径为4.6mm,柱粒度为3.5μm;色谱柱的温度为40℃;流动相为甲醇和0.8%冰乙酸的水溶液混合体系,混合体系中冰乙酸水溶液的体积分数为25%;每次进样的样品体积为5μl,流动相的流速为1ml/min,检测波长为280nm。结果如下表4所示,色谱峰的保留时间稳定,比较峰面积得到,rsd小于1%,表明本发明所述的分析方法稳定性良好。
[0087]
表4稳定性试验结果
[0088][0089]
试验例2精密度试验
[0090]
取实施例2小试样品20201208批的7
‑
氯茚并[1,2
‑
e][1,3,4]恶二嗪
‑
2,4a(3h,5h)
‑
二羧酸
‑
4a
‑
甲酯
‑2‑
苄酯为考察对象,称取标准品和五个平行待测样品,按照标准品溶液、待测样品溶液、待测样品溶液、标准品溶液溶液的顺序分别进样,计算五个平行待测样品中7
‑
氯茚并[1,2
‑
e][1,3,4]恶二嗪
‑
2,4a(3h,5h)
‑
二羧酸
‑
4a
‑
甲酯2
‑
苄酯的质量分数;
[0091]
分析条件为:采用高效液相色谱仪和二极管阵列检测器的岛津液相色谱仪,色谱柱为ods
‑
c18反相色谱柱,色谱柱的柱长为150mm,柱内径为4.6mm,柱粒度为3.5μm;色谱柱的温度为40℃;流动相为甲醇和0.8%冰乙酸水的混合体系,混合体系中冰乙酸水的体积分
数为25%;每次进样的样品体积为5μl,流动相的流速为1ml/min;检测波长设定为280nm。
[0092]
结果如下表5所示,比较7
‑
氯茚并[1,2
‑
e][1,3,4]恶二嗪
‑
2,4a(3h,5h)
‑
二羧酸
‑
4a
‑
甲酯
‑2‑
苄酯的质量分数得到,rsd小于1%,表明本发明所述的分析方法精密度良好。
[0093]
表5精密度试验结果
[0094][0095][0096]
试验例3回收率试验
[0097]
取实施例2小试样品20201208批7
‑
氯茚并[1,2
‑
e][1,3,4]恶二嗪
‑
2,4a(3h,5h)
‑
二羧酸
‑
4a
‑
甲酯
‑2‑
苄酯,称取标样,同时分别称取五个不同质量的样品,并加入不同质量的标样,进样计算加入标样的回收率。
[0098]
分析条件为:采用高效液相色谱仪带二极管阵列检测器,色谱柱为ods
‑
c18手性色谱柱,色谱柱的柱长为150mm,柱内径为4.6mm,柱粒度为3.5μm;色谱柱的温度为40℃;流动相为甲醇和0.8%冰乙酸水溶液的混合体系,混合体系中冰乙酸水溶液的体积分数为25%,每次进样的样品体积为5μl,流动相的流速为1ml/min;将高效液相色谱的检测波长设定为280nm。仪器基线稳定后按标准品、待测样品、标准品、待测样品的顺序依次进样,分别计算标准品溶液、待测样品溶液的7
‑
氯茚并[1,2
‑
e][1,3,4]恶二嗪
‑
2,4a(3h,5h)
‑
二羧酸
‑
4a
‑
甲酯
‑2‑
苄酯峰面积平均值,结果如下表6所示,回收率均在98%
‑
101%之间,平均回收率为99.38%,表明该实验回收率符合要求。
[0099]
表6回收率试验结果
[0100]
[0101][0102]
试验例4线性试验
[0103]
称取一系列不同质量的标准品,置于100ml容量瓶中,用甲醇溶解并定容,进样后考察峰面积和溶液浓度的关系;
[0104]
分析条件包括:采用高效液相色谱仪带二极管阵列检测器,色谱柱为ods
‑
c18反相色谱柱,色谱柱的柱长为150mm,柱内径为4.6mm,柱粒度为3.5μm;色谱柱的温度为40℃;流动相为甲醇和0.8%冰乙酸水溶液的混合体系,混合体系中冰乙酸水溶液的体积分数为25%;每次进样的样品体积为5μl,流动相的流速为1ml/min,检测波长为280nm。
[0105]
结果如下表7及图7所示,相关系数为1,表明本发明提供的分析方法线性符合要求。
[0106]
表7线性试验结果
[0107][0108][0109]
综合上述试验例1
‑
4可知,本发明所提供的7
‑
氯茚并[1,2
‑
e][1,3,4]恶二嗪
‑
2,4a(3h,5h)
‑
二羧酸
‑
4a
‑
甲酯
‑2‑
苄酯含量的分析方法准确性高,可操作性好,可以广泛地应用到7
‑
氯茚并[1,2
‑
e][1,3,4]恶二嗪
‑
2,4a(3h,5h)
‑
二羧酸
‑
4a
‑
甲酯
‑2‑
苄酯含量的分析检测中。
[0110]
对比例1不同检测波长下线性检测结果的对比
[0111]
在试验过程中,选择了260nm、300nm波长分别进行线性验证,结果见表8
‑
9。验证发现,在波长260nm、300nm条件下,虽然线性相关系数r2的值也大于0.99,但截距很大,参见图8与图9,因此如果采用本发明中的单标准比较法进行样品分析,误差较大。
[0112]
表8波长在260nm下线性试验结果
[0113][0114]
表9波长在300nm下线性试验结果
[0115][0116][0117]
对比例2不同流动相比例下色谱图情况
[0118]
取小试试样,以甲醇:0.8%乙酸水溶液=90:10(或60:40)的混合体系为流动相,流速为1ml/min,每次进样的样品体积为5μl,考察不同流动相比例对色谱图出峰情况。
[0119]
在甲醇:0.8%乙酸水溶液=90:10时,有些小的杂质与目标物分离度很差,影响峰面积的计算,进而影响含量计算,见图10。
[0120]
在甲醇:0.8%乙酸水溶液=60:40时,当有机相的配比降低后,出峰的时间延长并且峰型变宽,见图11。
[0121]
尽管通过参考附图并结合优选实施例的方式对本发明进行了详细描述,但本发明并不限于此。在不脱离本发明的精神和实质的前提下,本领域普通技术人员可以对本发明的实施例进行各种等效的修改或替换,而这些修改或替换都应在本发明的涵盖范围内任何熟悉本技术领域的技术人员在本发明揭露的技术范围内,可轻易想到变化或替换,都应涵盖在本发明的保护范围之内。因此,本发明的保护范围应以权利要求所述的保护范围为准。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1