一种检测体内精神科常用药物或其代谢物的方法和应用与流程
1.本发明涉及精神科药物血药浓度的检测领域,具体涉及一种检测体内精神科常用药物或其代谢物的方法和应用。
背景技术:
2.抗精神病药物(antipsychotic drugs)又称强安定药或神经阻滞剂(neuroleptic),是一组用于治疗精神分裂症及其它精神病性精神障碍的药物。在通常的治疗剂量并不影响患者的智力和意识,却能有效地控制患者的精神运动兴奋、幻觉、妄想、敌对情绪、思维障碍和异常行为等精神症状。病人通常存在睡眠障碍,常常同时服用镇静安神类药物,为此需要在检测时将二者分离,以获得稳定的抗精神病药物的血药浓度检测值,避免其余临床合并用药的干扰。精神科病人常用的临床治疗药物有:奥氮平、9-oh利培酮、氯氮平、去甲氯氮平、利培酮、富马酸喹硫平、盐酸氯丙嗪、盐酸曲唑酮、米氮平、氨磺必利、阿立哌唑、脱氢阿立哌唑,而为了配合治疗,病人通常也会合并使用如阿普唑仑、氯硝西泮、佐匹克隆、艾司唑仑等镇静安眠类药物,如果检测方法种不能实现同时分离此类合并用药,会导致精神科药物浓度定量不准,因此将镇静安眠类药物与抗精神病药物同时实现在线分离具有非常重要的临床应用价值。
3.治疗药物浓度监测是指在临床进行的药物治疗中,在指定的时间间隔测量患者血液中特定药物的浓度。现阶段国内临床血药浓度监测的方法:主要有高效液相色谱法(hplc)、放射免疫法(ria)、气相色谱法(gc)和液相串联质谱法(lc-ms)等。由于临床病人常常同时服用多种精神基本疾病类药物,高效液相色谱紫外检测方法检测时经常不能完全实现药物的色谱分离,容易存在干扰,影响定量结果;高效液相色谱串联质谱法是以质谱仪为检测手段,集高效液相色谱仪的高分离能力、质谱仪的高灵敏度和高选择性于一体的强有力分析检测工具,质谱作为检测手段虽可以准确定量,但是一方面设备较贵,另一方面同位素内标耗材也非常昂贵且对环境不友好。因此,本领域亟需一种将以上两类药物同时实现在线分离、且分离时间短、效果好的检测方法。
技术实现要素:
4.为克服现有技术缺陷,本发明采用了以下技术方案:
5.本发明第一方面提供了一种同时检测生物样本中精神科常用药物或其代谢物的方法,其特征在于,所述方法为采用超高效液相色谱-紫外检测法,包括以下步骤:
6.s1色谱条件:(1)色谱柱:c
18
色谱柱;(2)流动相:a相为甲酸-水溶液,b相为甲酸-甲醇溶液;(3)检测器为紫外(uv)检测器;
7.s2工作溶液的配制:
8.s21内标溶液的制备:
9.s211胃复安内标工作溶液:精密吸取适量胃复安标准品,先用少量甲醇溶解,再用甲醇-水溶液定容,配制成胃复安标准品溶液,再用甲醇-水溶液稀释定容,即得内标1-胃复
安工作溶液;
10.s212卡马西平内标工作溶液:精密吸取适量卡马西平标准品,先用少量甲醇溶解,再用甲醇-水溶液定容,配制成卡马西平标准品溶液,再用甲醇-水溶液稀释至定容,即得内标2-卡马西平工作溶液;
11.s22混合标准品溶液的制备:
12.s221混标母液的配制:分别精密称定适量神科常用药物或其代谢物氨磺必利、氯氮平、去甲氯氮平、阿立哌唑、脱氢阿立哌唑、奥氮平、米氮平、利培酮、9-oh利培酮、佐匹克隆、曲唑酮、氯丙嗪、喹硫平、氯硝西泮、艾司唑仑以及阿普唑仑,混合后先用适量溶剂溶解后,再采用甲醇-水溶液稀释定容配制成混标母液;
13.s222定性混合标准品溶液的制备:
14.将上述混标母液用甲醇-水溶液按一定比例稀释,配制成含氨磺必利、氯氮平、去甲氯氮平、阿立哌唑、脱氢阿立哌唑浓度均为80μg/ml,含奥氮平、米氮平、利培酮、9-oh利培酮、佐匹克隆浓度均为20μg/ml,含曲唑酮、氯丙嗪、喹硫平浓度为40μg/ml,以及含氯硝西泮、艾司唑仑、阿普唑仑浓度均为1.0μg/ml的定性混合标准品溶液;
15.s223混合标准品溶液的制备:分别精密吸取上述定性混合标准品溶液10~40μl,然后加入上述内标1-胃复安工作溶液以及内标2-卡马西平工作溶液各10~40μl,干燥后加入适量空白血液、血浆、唾液或血清以及甲基叔丁基醚,第一次涡旋,然后加入100~500μl的浓度为1~3mol/l的氢氧化钠水溶液以及甲基叔丁基醚第二次涡旋并离心,获得上清液,将所述上清液干燥后再加入含甲酸的甲醇-水溶液复溶、涡旋、过滤,即得;
16.s23供试品溶液的制备包括:
17.s231供试品溶液的制备:分别精密吸取s21制备的内标1-胃复安工作溶液以及内标2-卡马西平工作溶液各10~40μl,干燥后与适量待测血液、血浆、唾液或血清混合,第一次涡旋,加入100~500μl氢氧化钠水溶液以及甲基叔丁基醚进行第二次涡旋,涡旋后离心获得上清液,取上清液干燥后再加入含甲酸的甲醇-水溶液复溶、涡旋、过滤即得;
18.或s232头发供试品溶液的制备:分别精密吸取s21制备的内标1-胃复安工作溶液以及内标2-卡马西平工作溶液各10~40μl,干燥后与适量待测头发样本混合,第一次涡旋,加入100~500μl氢氧化钠水溶液,超声或静置,然后加入甲基叔丁基醚进行第二次涡旋,涡旋后离心获得上清液,取上清液干燥后加入含甲酸的甲醇-水溶液复溶、涡旋、过滤,即得;
19.s3测定法:将s22制备的混合标准品溶液以及s23制备的供试品溶液分别注入超高效液相色谱仪,采用梯度洗脱程序,得到色谱图,获得所述精神类药物及其代谢物的保留时间tr,即得;
20.进一步的,所述生物样本选自血液、血浆、血清、唾液、头发中的任意一种或几种;进一步优选的,所述生物样本选自血浆、血清、头发中的任意一种或几种;
21.进一步的,s1所述的色谱条件中:流动相:a相为0.05v/v%甲酸-水溶液;且b相为0.05v/v%甲酸-甲醇溶液;
22.进一步的,s1所述的色谱条件中:紫外检测器为vwd或dad检测器;进一步优选的,所述紫外检测器的检测波长为254nm、285nm或全波长扫描;
23.更进一步的,s1所述的色谱条件还包括:(4)流动相流速为0.1~0.5ml/min;(5)柱温为35~45℃;(6)工作溶液的进样量均为1.0~20.0μl。
24.进一步的,步骤s21中,所述内标溶液的制备具体包括:
25.s211内标1-胃复安工作溶液:精密称定1~30mg胃复安标准品,先用1~30ml甲醇溶解,再用40~70%v/v甲醇-水溶液定容,配制成浓度为0.1~0.5mg/ml的胃复安储备溶液,再用40~70%v/v甲醇-水溶液稀释至10~40μg/ml,即得内标1-胃复安工作溶液;
26.s212内标2-卡马西平工作溶液:精密称定1~30mg卡马西平标准品,先用1~30ml甲醇溶解,再用40~70%v/v甲醇-水溶液定容,配制成浓度为0.1~0.5mg/ml的卡马西平储备溶液,再用40~70%v/v甲醇-水溶液稀释至1~5μg/ml,即得内标2-卡马西平工作溶液;
27.进一步的,所述s22混合标准品溶液的制备具体包括:
28.s221混标母液的配制:分别精密称定适量神科常用药物或其代谢物氨磺必利、氯氮平、去甲氯氮平、阿立哌唑、脱氢阿立哌唑、奥氮平、米氮平、利培酮、9-oh利培酮、佐匹克隆、曲唑酮、氯丙嗪、喹硫平、氯硝西泮、艾司唑仑以及阿普唑仑1~30mg,混合后先用1~30ml溶剂溶解后,再采用40~70%v/v甲醇-水溶液稀释定容配制成浓度为0.1~0.5mg/ml混标母液;
29.s222定性混合标准品溶液的制备:将上述混标母液用40~70v/v%甲醇-水溶液按一定比例稀释,配制成含氨磺必利、氯氮平、去甲氯氮平、阿立哌唑、脱氢阿立哌唑浓度为80μg/ml,含奥氮平、米氮平、利培酮、9-oh利培酮、佐匹克隆浓度为20μg/ml,含曲唑酮、氯丙嗪、喹硫平浓度为40μg/ml,以及含氯硝西泮、艾司唑仑、阿普唑仑浓度为1.0μg/ml的定性混合标准品溶液;
30.s223混合标准品溶液的制备:分别精密吸取上述定性混合标准品溶液10~40μl至6~10ml离心管中,然后加入上述内标1-胃复安工作溶液以及内标2-卡马西平工作溶液各10~40μl,氮气吹干后加入400~1000μl空白血清以及甲基叔丁基醚2~7ml,第一次涡旋0.5~2min,然后加入100-500μl浓度为1~3mol/l的氢氧化钠水溶液以及甲基叔丁基醚2~7ml,第二次涡旋2~5min,再离心4~10min,取离心后的上清液,氮气吹干后加入60~250μl含0.05~0.15v/v%甲酸、浓度为10~20%v/v甲醇-水溶液复溶,涡旋0.5~2min,滤膜过滤,即得;进一步优选的,分别精密吸取上述定性混合标准品溶液20μl至7ml离心管中,然后加入上述内标1-胃复安工作溶液以及内标2-卡马西平工作溶液各20μl,氮气吹干后加入1000μl空白血液、血浆、唾液或血清以及甲基叔丁基醚3ml,第一次涡旋1min,然后加入200μl浓度为2mol/l的氢氧化钠水溶液以及甲基叔丁基醚3ml,第二次涡旋3min,再离心5min,取离心后的上清液,氮气吹干后加入200μl含0.1v/v%甲酸、浓度为15%v/v甲醇-水溶液复溶,涡旋1min,滤膜过滤,即得;
31.进一步的,所述s231血液、血浆、唾液或血清供试品溶液的制备包括以下步骤:分别精密吸取s21制备的内标1-胃复安工作溶液以及内标2-卡马西平工作溶液各10~40μl,氮气吹干后与400~1000μl融化后的待测冷冻血液、血浆、唾液或血清样品混合至7~10ml离心管中,第一次涡旋0.5~2min,加入100~500μl浓度为1~3mol/l的氢氧化钠水溶液,以及甲基叔丁基醚2~7ml,再进行第二次涡旋2~5min,然后将涡旋后离心管以速度为2500~5000转/分离心4~10min,取离心后的上清液氮气吹干后加入60~250μl含0.05~0.15v/v%甲酸、且浓度为10~20v/v%甲醇-水溶液复溶,涡旋0.5~2min,尼龙滤膜过滤,即得;进一步优选的,分别精密吸取s21制备的内标1-胃复安工作溶液以及内标2-卡马西平工作溶液各20μl,氮气吹干后与1000μl融化后的待测冷冻血液、血浆、唾液或血清样品混合至7ml
离心管中,第一次涡旋1min,加入200μl浓度为2mol/l氢氧化钠水溶液,加入甲基叔丁基醚3ml,第二次涡旋3min,再离心5min,取离心后的上清液,氮气吹干后加入200μl含0.1v/v%甲酸、浓度为15%v/v甲醇-水溶液复溶,涡旋1min,滤膜过滤,即得;
32.或进一步的,所述s232头发供试品溶液的制备包括以下步骤:取待测头发用丙酮、水依次清洗,干燥后剪碎,分别精密吸取s21制备的内标1-胃复安工作溶液以及内标2-卡马西平工作溶液各10~40μl,氮气吹干后与10~50mg待测头发样品混合至7~10ml离心管中,加入100~500μl浓度为1~3mol/l的氢氧化钠水溶液,第一次涡旋0.5~2min,然后超声1~4h,再加入甲基叔丁基醚2~7ml,再进行第二次涡旋2~5min,将涡旋后离心管以速度为2500~5000转/分离心4~10min,取离心后的上清液氮气吹干后加入60~250μl含0.05~0.15v/v%甲酸、且浓度为10~20v/v%甲醇-水溶液复溶,涡旋0.5~2min,尼龙滤膜过滤,即得;进一步优选的,取待测头发用丙酮、水依次清洗,干燥后剪碎,分别精密吸取s21制备的内标1-胃复安工作溶液以及内标2-卡马西平工作溶液各20μl,氮气吹干后与20mg待测头发样品混合至7ml离心管中,加入200μl浓度为2mol/l的氢氧化钠水溶液,第一次涡旋1min,然后超声2h,再加入甲基叔丁基醚4ml,再进行第二次涡旋3min,将涡旋后离心管以速度为3000转/分离心5min,取离心后的上清液氮气吹干后加入200μl含0.1v/v%甲酸、且浓度为15v/v%甲醇-水溶液复溶,涡旋1min,尼龙滤膜过滤,即得;
33.进一步的,所述s3测定法中所述的梯度洗脱程序为:0min 5%v/v b、2min 10%v/v b、4min 20%v/v b、6min 22%v/v b、12min 35%v/v b、15min 36%v/v b、18.5min 40%v/v b、20min 50%v/v b、21min 90%v/v b、23min 90%v/v b、24min 5%v/v b以及28min 5%v/v b;
34.本发明的第二方面提供了上述任一项检测方法在制备生物样本中精神类药物或其代谢物检测试剂中的应用。
附图说明
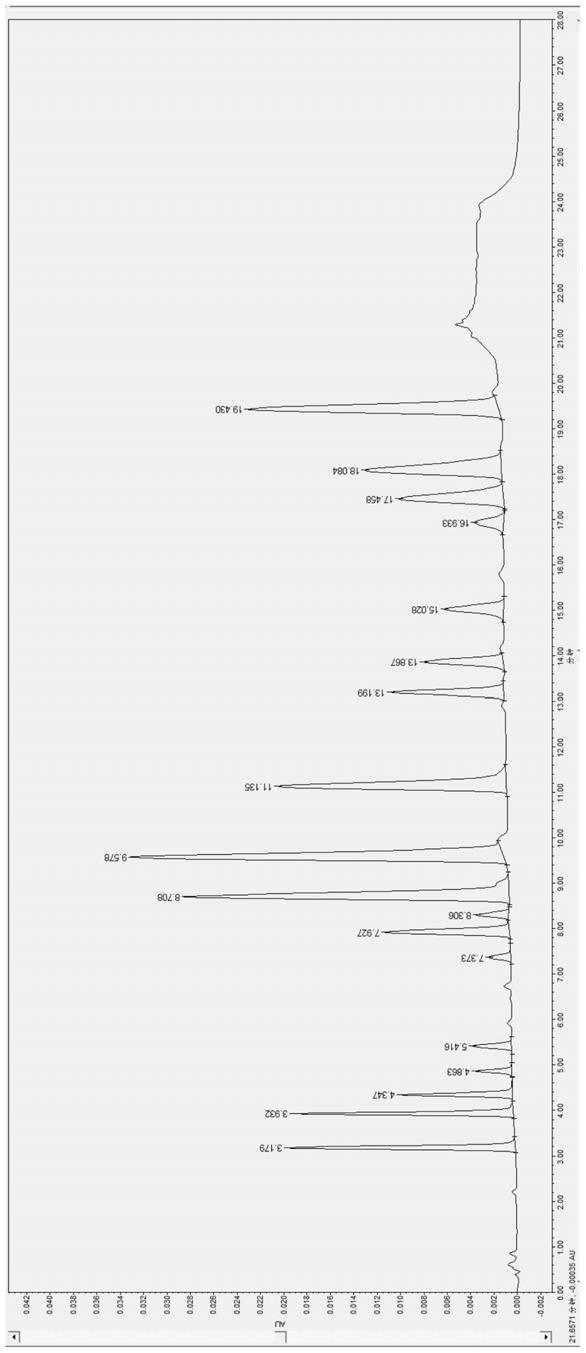
35.图1为实施例1注入标准品溶液的色谱图
36.图2为实施例2服用氯氮平、富马酸喹硫平药物的病人血清样品的色谱图
37.图3为实施例3服氨磺必利药物的病人血清样品的检测色谱图
38.图4为实施例4服用氯氮平药物的病人血清样品的检测色谱图
39.图5为实施例5服用阿立哌唑药物的病人血清样品的检测色谱图
40.图6为实施例6服用奥氮平、曲唑酮、利培酮药物的病人头发样品的检测色谱图
41.图7为实施例7服用奥氮平、米氮平药物的病人头发样品的检测色谱图
42.图8为实施例8服用喹硫平药物病人头发样品的检测色谱图
43.有益效果
44.1.本发明采用uplc-uv法实现了人血清或头发中精神科常用药物及部分活性代谢物的分离;
45.2.本发明采用双内标的分析方法,可以实现快速准确检测,通过向标准品溶液和供试品溶液中加入2个内标物质(胃复安、卡马西平)同时进样,以准确定位不同保留时间样品峰峰位,若一种内标干扰,则可采用另一种物质作为内标,仅采用不到30min就实现了多种检测药物的分离,大大缩短了检测时间;
46.3.本发明采用较为经济的紫外检测器替代使用质谱进行检测,对于指导临床用药以及分析样本具有显著地学术意义和经济价值;
47.4.本发明通过进一步优化分离条件,例如增加流速、优化洗脱梯度程序,实现短时间内16种药物同时在线分离;
48.5.本发明采用液-液萃取法、通过加入naoh溶液提取生物样本(特别是头发)中的有效成分,显著提高了检测限度,且无须昂贵的固相萃取耗材,过滤后进样使色谱柱更耐用,适用于长期大量样本临床检测,具有显著地经济价值;
49.6.本发明选择特定的复溶液复溶,进一步提高了测量的准确度。
50.7.本发明进一步优选了过滤滤膜的材质,避免过滤造成损失。
具体实施方式
51.实施例1:完整的混合标准品的检测方法
52.一种同时检测体内精神类药物或其代谢物的方法,所述方法为采用超高效液相色谱-紫外检测法,包括以下步骤:
53.s1色谱条件:(1)色谱柱:c
18
色谱柱,acquity uplc beh c
18
直径2.1mm*50mm,其填料粒径为1.7μm;(2)流动相:a相为0.05v/v%甲酸-水溶液;b相为0.05v/v%甲酸-甲醇溶液;(3)检测器为紫外(uv)检测器,所述紫外检测器的检测波长全波长扫描;(4)流动相流速为0.4ml/min;(5)柱温为40℃;(6)工作溶液的进样量均为10.0μl;
54.s2工作溶液的配制包括:
55.s20洗针溶液配制:用量筒量取700ml水至液相洗瓶中,再加入300ml甲醇,混合超声20min后,制得甲醇-水洗针溶液;
56.s21内标溶液的制备:
57.s211内标1-胃复安工作溶液:精密称定20mg胃复安标准品于100ml容量瓶中,先用10ml甲醇溶解,再用50%v/v甲醇-水溶液定容,配制成浓度为0.2mg/ml的胃复安储备溶液,再用50%v/v甲醇-水溶液稀释至20μg/ml,即得内标1-胃复安工作溶液;
58.s212内标2-卡马西平工作溶液:精密称定20mg卡马西平标准品,先用10ml甲醇溶解于100ml容量瓶中,再用50%v/v甲醇-水溶液定容,配制成浓度为0.2mg/ml的卡马西平储备溶液,再用50%v/v甲醇-水溶液稀释至2μg/ml,即得内标2-卡马西平工作溶液;
59.s22混合标准品溶液的制备:
60.s221混标母液的配制:分别精密称定各精神疾病类药物或其代谢物氨磺必利、氯氮平、去甲氯氮平、阿立哌唑、脱氢阿立哌唑、奥氮平、米氮平、利培酮、9-oh利培酮、佐匹克隆、曲唑酮、氯丙嗪、喹硫平、氯硝西泮、艾司唑仑以及阿普唑仑标准品20mg于100ml容量瓶中,除阿立哌唑、脱氢阿立哌唑外其余药物用10ml甲醇溶解,阿立哌唑、脱氢阿立哌唑用含1%甲酸的10ml甲醇溶液溶解,完全溶解后,再用50%v/v甲醇-水溶液定容,配制成浓度为0.2mg/ml的标准品母液;
61.s222定性混合标准品溶液的配制:将上述混标母液用50v/v%甲醇-水溶液按一定比例稀释,配制成含氨磺必利、氯氮平、去甲氯氮平、阿立哌唑、脱氢阿立哌唑浓度为80μg/ml,含奥氮平、米氮平、利培酮、9-oh利培酮、佐匹克隆浓度为20μg/ml,含曲唑酮、氯丙嗪、喹硫平浓度为40μg/ml,以及含氯硝西泮、艾司唑仑、阿普唑仑浓度为1.0μg/ml的定性混合标
准品溶液;
62.s223混合标准品溶液的制备:分别精密吸取上述定性混合标准品溶液20μl至7ml离心管中,然后加入上述内标1-胃复安工作溶液以及内标2-卡马西平工作溶液各20μl,氮气吹干后加入1000μl空白血清以及甲基叔丁基醚3ml,第一次涡旋1min,然后加入200μl浓度为2mol/l的氢氧化钠水溶液以及甲基叔丁基醚3ml,第二次涡旋3min,再离心5min,取离心后的上清液,氮气吹干后加入200μl含0.1v/v%甲酸、浓度为15%v/v甲醇-水溶液复溶,涡旋1min,滤膜过滤,即得;
63.s23供试品溶液的制备包括:
64.s231血清供试品溶液的制备:分别精密吸取s21制备的内标1-胃复安工作溶液以及内标2-卡马西平工作溶液各20μl,氮气吹干后与1000μl融化后的待测冷冻血清样品混合至7ml离心管中,第一次涡旋1min,加入200μl浓度为2mol/l氢氧化钠水溶液,加入甲基叔丁基醚3ml,第二次涡旋3min,再离心5min,取离心后的上清液,氮气吹干后加入200μl含0.1v/v%甲酸、浓度为15%v/v甲醇-水溶液复溶,涡旋1min,滤膜过滤,即得;
65.s3测定法:将混合标准品溶液注入超高效液相色谱仪,采用如下梯度洗脱程序:0min 5%v/v b、2min 10%v/v b、4min 20%v/v b、6min22%v/v b、12min 35%v/v b、15min 36%v/v b、18.5min 40%v/v b、20min 50%v/v b、21min 90%v/v b、23min 90%v/v b、24min 5%v/v b以及28min 5%v/v b;得到定性混合标准品工作溶液色谱图如图1所示,获得所述精神药物及其代谢物保留时间tr,即得;所述的精神药物及其代谢物保留时间tr分别为奥氮平3.179min、氨磺必利3.932min、胃复安4.347(内标1)、佐匹克隆4.863min、米氮平5.416min、9-oh利培酮7.373min、曲唑酮7.927min,利培酮8.306min、去甲氯氮平8.708min、氯氮平9.578min、喹硫平11.135min、卡马西平13.199min(内标2)、氯硝西泮、艾司唑仑、阿普唑仑、脱氢阿立哌唑17.397min、阿立哌唑18.035min、氯丙嗪19.419min。
66.实施例2服用氯氮平、富马酸喹硫平药物的病人血清样品的检测
67.一种同时检测体内精神类药物或其代谢物的方法,所述方法为采用超高效液相色谱-紫外检测法,包括以下步骤:
68.s1色谱条件、s21内标溶液、s22混合标准品溶液的制备方法同实施例1;
69.s231血清供试品溶液制备:分别精密吸取s21制备的内标1-胃复安工作溶液以及内标2-卡马西平工作溶液各20μl,氮气吹干后与500μl融化后的待测冷冻血清样品混合至7ml离心管中,第一次涡旋1min,加入200μl浓度为2mol/l的氢氧化钠水溶液,以及甲基叔丁基醚3ml,再进行第二次涡旋3min,然后将涡旋后离心管以速度为3000转/分离心5min,取离心后的上清液氮气吹干后加入150μl含0.1v/v%甲酸、且浓度为15v/v%甲醇-水溶液复溶,涡旋1min,用0.25μm尼龙滤膜过滤,即得;
70.s3测定法:将供试品溶液注入超高效液相色谱仪,采用梯度洗脱程序(同实施例1),得到色谱图,如图2所示,获得所述精神药物及其代谢物保留时间tr分别为:内标1胃复安4.348min;去甲氯氮平8.897min;氯氮平9.783min;喹硫平11.262min;内标2卡马西平13.206min。
71.实施例3服氨磺必利病人的血清样品的检测
72.s1色谱条件、s21内标溶液、s22混合标准品溶液的制备方法同实施例1;
73.s231供试品溶液制备、s3测定法同实施例2:
74.结果如图3所示,获得所述精神药物及其代谢物保留时间tr,获得所述精神药物及其代谢物保留时间tr分别为:氨磺必利的保留时间tr为3.949min;内标1胃复安4.361min;内标2卡马西平13.218min。
75.实施例4服用药物氯氮平病人的血清样品的检测
76.s1色谱条件、s21内标溶液、s22混合标准品溶液的制备方法同实施例1;
77.s231供试品溶液制备、s3测定法同实施例2;
78.结果:如图4所示,获得所述精神药物及其代谢物保留时间tr,获得所述精神药物及其代谢物保留时间tr分别为:内标1胃复安4.354min;去甲氯氮平8.896min;氯氮平9.769min;内标2卡马西平13.203min。
79.实施例5服用药物阿立哌唑病人的血清样品的检测
80.s1色谱条件、s21内标溶液、s22混合标准品溶液的制备方法同实施例1;
81.s231供试品溶液制备、s3测定法同实施例2;
82.结果:如图5所示,获得所述精神药物及其代谢物保留时间tr分别为:内标1胃复安4.363min;内标2卡马西平13.216min;脱氢阿立哌唑17.614min;阿立哌唑18.208min。
83.实施例6服用奥氮平、曲唑酮、利培酮的病人头发样品的检测
84.s1色谱条件、s21内标溶液、s22混合标准品溶液的制备方法同实施例1;
85.s231供试品溶液制备、s3测定法同实施例2;
86.s232头发供试品溶液的制备包括以下步骤:
87.贴头皮剪取患者枕后部头发一搓,用丙酮、水依次清洗,取贴近头皮1cm的一段干燥后剪碎待用,精密称量待测头发20.45mg至7ml离心管中,分别精密吸取s21制备的内标1-胃复安工作溶液以及内标2-卡马西平工作溶液各20μl,氮气吹干,加入200μl浓度为2mol/l的氢氧化钠水溶液,第一次涡旋1min,然后超声2.5h,再加入甲基叔丁基醚3ml,再进行第二次涡旋3min,将涡旋后离心管以速度为3000转/分离心5min,取离心后的上清液氮气吹干后加入200μl含0.1v/v%甲酸、且浓度为15v/v%甲醇-水溶液复溶,涡旋1min,用0.25μm尼龙滤膜过滤,即得;
88.结果:如图6所示,获得所述精神药物及其代谢物保留时间tr分别为:内标1胃复安的保留时间tr为4.367min;9-oh利培酮tr为7.209min;利培酮的保留时间tr为8.440min;喹硫平的保留时间tr为11.23min,内标2卡马西平的保留时间tr为13.203min。
89.实施例7服用奥氮平、米氮平药物的病人头发样品的检测
90.s1色谱条件、s21内标溶液、s22混合标准品溶液的制备方法同实施例1;s232头发供试品溶液的制备方法同实施例6;s3测定法同实施例2;记录实际称量的待测头发质量为20.35mg。
91.结果:如图7所示,所测得的精神药物及其代谢物保留时间tr分别为奥氮平3.200min;内标1胃复安4.367min;米氮平5.397min,内标2卡马西平13.217min。
92.实施例8:服用喹硫平药物病人头发样品的检测
93.s1色谱条件、s21内标溶液、s22混合标准品溶液的制备方法同实施例1;s232头发供试品溶液的制备方法同实施例5;s3测定法同实施例6;记录实际称量的待测头发质量为20.45mg。
94.结果:如图8所示,获得所述精神药物及其代谢物保留时间tr,分别为内标1胃复安
4.367min;喹硫平11.230min;内标2卡马西平13.211min。
95.虽然本发明已以较佳实施例公开如上,但其并非用以限定本发明,任何熟悉此技术的人,在不脱离本发明的精神和范围内,都可做各种的改动与修饰,均在本发明的保护范围内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1