一种检测草甘膦的方法
1.本发明属于农药检测技术领域,尤其涉及一种检测草甘膦的方法。
背景技术:
2.草甘膦是一种内吸传导型广谱灭生性除草剂,对多年生根杂草非常有效,广泛用于橡胶、桑、茶、果园及甘蔗地,因其较好的效果在全球销量位居前几位。草甘膦的大量使用导致其成为在农产品中易检出的农药,研究表明,在土壤和饮用水中草甘膦残留都有检出,并且可通过水生环境进入食物链,同时其环境稳定性高,对人体健康构成威胁。草甘膦不可逆地灭活乙酰胆碱酯酶,可引起不同的生理反应,如呼吸、心肌和神经肌肉功能障碍,因此,迫切需要开发灵敏、便捷的分析方法来识别环境和农产品中残留的草甘膦。目前我国农产品中草甘膦残留的标准检测方法以液相色谱-质谱/质谱联用法为主,同时,还可用气相色谱衍生化、液相色谱衍生化方法,但这些方法所需要的仪器昂贵、过程耗时较长(在30min以上)、成本较高,限制了其这些技术的推广使用,因此,有必要针对快速检测草甘膦残留的需求,开发合适的草甘膦残留快速检测方法。
技术实现要素:
3.有鉴于此,本发明的目的在于提供一种检测草甘膦的方法,采用本技术提供的方法,5min得到样品中草甘膦的浓度,缩短了检测草甘膦的时间。
4.为了实现上述发明目的,本发明提供了以下技术方案:
5.本发明提供了一种检测草甘膦的方法,包括以下步骤:
6.1)将样品与含铈离子溶液、焦磷酸钠溶液混合,得到混合液;
7.2)将所述步骤1)得到的混合液振荡,得到振荡液;
8.3)测定所述步骤2)得到的振荡液的荧光强度,将所述荧光强度代入线性方程,得到样品中草甘膦的浓度;
9.所述线性方程为:f=-4097.888logc+9356.104,c的单位为μm/l。
10.优选的,所述步骤1)样品包括自来水。
11.优选的,所述步骤1)含铈离子溶液包括硝酸铈溶液。
12.优选的,所述硝酸铈溶液的浓度为0.25m~1m。
13.优选的,所述步骤1)焦磷酸钠溶液的浓度为1mm。
14.优选的,所述步骤1)样品与含铈离子溶液、焦磷酸钠溶液的体积比为3:8~10:2。
15.优选的,所述步骤2)振荡的时间为5min,振荡的转速为600rpm。
16.优选的,所述步骤3)测定的条件包括:在300nm激发下记录346nm处的荧光强度。
17.本发明提供了一种检测草甘膦的方法,包括以下步骤:1)将样品与含铈离子溶液、焦磷酸钠溶液混合,得到混合液;2)将所述步骤1)得到的混合液振荡,得到振荡液;3)测定所述步骤2)得到的振荡液的荧光强度,将所述荧光强度代入线性方程,得到样品中草甘膦的浓度;所述线性方程为:f=-4097.888logc+9356.104,c的单位为μm/l。
18.本发明检测草甘膦的机理是:
19.草甘膦与ce
3+
配位形成草甘膦-ce
3+-焦磷酸根(ppi)复合物,干扰ppi的配体场效应(这一效应可使ce
3+
荧光增强),降低荧光。
20.本发明的有益效果:采用本发明提供的检测草甘膦的方法,检测时间仅需5min,检测限为2.37μg/l(0.014μm)。
附图说明
21.图1为实施例1的ce-ppi的扫描电镜结果;
22.图2为对材料进行元素分析的结果,a图是6μm尺度下的电镜图,b图和c图分别是p元素和ce元素的元素分析结果图;
23.图3为实施例1的ce-ppi的x射线光电子能谱结果;
24.图4为实施例1的ce-ppi的荧光光谱结果;
25.图5为实施例1的ce-ppi的吸收光谱结果;
26.图6为草甘膦对ce-ppi荧光性质的影响;
27.图7为实施例3ce-ppi荧光检测草甘膦结果;
28.图8为根据线性方程做的0.1-5μm的标准曲线图;
29.图9为不同时间点下的相对荧光强度对时间做图,保持草甘膦与材料结合时间为15min,使得ppi与ce3+结合时间分别为0、5、10、15、20min,以每个时间点下的相对荧光强度对时间做图,观察变化,找到最佳结合时间;
30.图10为不同时间点下的相对荧光强度对时间做图,保持草甘膦与材料结合时间为5min,使得ppi与ce3+结合时间分别为0、5、10、15、20min,以每个时间点下的相对荧光强度对时间做图,观察变化,找到最佳结合时间。
具体实施方式
31.本发明提供了一种检测草甘膦的方法,包括以下步骤:
32.1)将样品与含铈离子溶液、焦磷酸钠溶液混合,得到混合液;
33.2)将所述步骤1)得到的混合液振荡,得到振荡液;
34.3)测定所述步骤2)得到的振荡液的荧光强度,将所述荧光强度代入线性方程,得到样品中草甘膦的浓度;
35.所述线性方程为:f=-4097.888logc+9356.104,c的单位为μm/l。
36.本发明将样品与含铈离子溶液、焦磷酸钠溶液混合,得到混合液。
37.在本发明中,所述样品优选包括自来水。在本发明中,当所述样品为自来水时,所述自来水经过滤膜后,再进行检测,所述滤膜的孔径优选为0.45μm。在本发明中,所述含铈离子溶液优选包括硝酸铈溶液。在本发明中,所述硝酸铈优选为六水合硝酸铈。在本发明中,所述硝酸铈溶液的浓度优选为0.25~1mm,更优选为0.5mm。在本发明中,所述焦磷酸钠溶液的浓度优选为1mm。在本发明中,所述样品与含铈离子溶液、焦磷酸钠溶液的体积比优选为3:8~10:2,具体为3:8:2、3:9:2和3:10:2。
38.本发明将得到的混合液振荡,得到振荡液。在本发明中,所述振荡的时间优选为5min,所述振荡的转速优选为600rpm。在本发明中,所述振荡的目的是使得不同组分充分混
合作用,达到荧光稳定。
39.本发明测定得到的振荡液的荧光强度,将所述荧光强度代入线性方程,得到样品中草甘膦的浓度;所述线性方程为:f=-4097.888logc+9356.104,c的单位为μm/l。
40.在本发明中,所述线性方程优选通过以下步骤得到:
41.通过将6μl不同浓度(10、25、50、100、250、500μm)的草甘膦溶液与20μl ce
3+
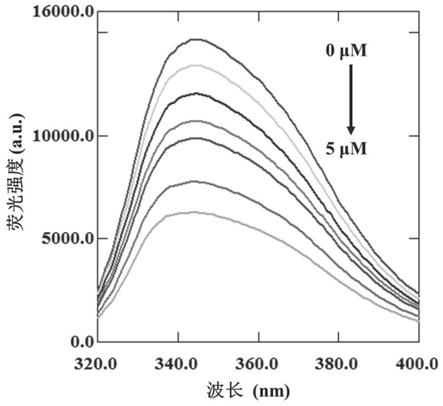
溶液(0.5mm)和4μl ppi溶液混合,将混合物在室温下以600rpm振荡5分钟。在300nm激发下记录荧光光谱和346nm处的荧光强度。重复该操作,得到不同浓度草甘膦下的荧光强度(每个浓度重复3次并取平均值,求标准偏差),以荧光强度对浓度的对数值做图即可得到线性方程。
42.在本发明中,所述线性方程为:f=-4097.888logc+9356.104,c的单位为μm/l,r2=0.9972,检测限为2.37μg/l(即0.014μm),检测草甘膦的线性范围:0.1~5μm。
43.下面结合实施例对本发明提供的技术方案进行详细的说明,但是不能把它们理解为对本发明保护范围的限定。
44.实施例1
45.铈离子-焦磷酸根配位聚合物网络(简称ce-ppi)的制备:
46.使用超纯水分别配制浓度为50mm的六水硝酸铈溶液和焦磷酸钠水溶液。将2ml六水合硝酸铈溶液和2ml焦磷酸钠水溶液在10ml塑料离心管中快速混合,1500rpm涡旋5min,产生的白色沉淀就是ce-ppi。得到的悬浮液以15000rpm离心10min,弃去上清液,白色沉淀用超纯水洗涤,重复上述操作3次后冷冻干燥得到ce-ppi。
47.对得到的ce-ppi扫描电镜(30kv),结果见图1。从图1中可以得出,铈离子-焦磷酸根配位聚合物网络的结构为球状纳米节点组成的网络结构,节点粒径为10~25nm。
48.ce-ppi的扫描电镜+元素分布结果见图2。
49.ce-ppi的x射线光电子能谱(xps)结果见图3,从图3中可以得出,ce-ppi的物性参:xps为:铈(3d3/2)904.4ev、铈(3d5/2)885.5ev、氧1s 531.3ev、磷2p 133.8ev。(xps条件:美国热电thermo escalab 250xi,相关参数:单色al ka(hv=1486.6ev),功率150w,500μm束斑,电荷校正采用污染碳c1s=284.8ev进行校正。a constant analyzer pass energy ep通能窄扫20ev,宽扫100ev,真空度1*10-10mba。能谱条件:horiba ex250日本堀场horiba产地日本)
50.ce-ppi的荧光光谱结果见图4,从图4中可以得出,最大激发波长:300nm,最大发射波长:346nm。
51.ce-ppi的吸收光谱结果见图5,从图5中可以得出,吸收峰约300nm。
52.实施例2
53.草甘膦对实施例1的ce-ppi荧光性质的影响:6μl草甘膦溶液(500μm)与20μl ce
3+
溶液(0.5mm)和4μl ppi溶液混合,然后将混合物在室温下以600rpm振荡5min,在300nm激发下记录荧光光谱和346nm处的荧光强度。
54.荧光光谱结果见图6,从图6中可以得出,草甘膦明显降低ce-ppi的荧光强度。
55.实施例3
56.基于实施例1的ce-ppi荧光检测草甘膦:6μl草甘膦溶液与20μl ce
3+
溶液(0.5mm)和4μl ppi溶液混合,然后将混合物在室温下以600rpm振荡5分钟,在300nm激发下记录荧光光谱和346nm处的荧光强度。
57.荧光光谱结果见图7,从图7中可以得出,ce-ppi荧光强度随草甘膦浓度的变化而产生的变化。
58.荧光强度与草甘膦浓度对数间的线性方程(标准方程,图8为标准曲线):f=-4097.888logc+9356.104,r2=0.9972,f代表荧光强度,c代表草甘膦的浓度,单位为μm/l。
59.草甘膦的浓度线性范围:0.1-5μm,检测限为2.37μg l-1(0.014μm)。
60.实施例4
61.检测自来水中的草甘膦浓度:
62.采用标准加入法进行,即使用一定体积的自来水配置成相应浓度草甘膦溶液,溶液过滤膜后进行检测。
63.样品详细配制方法:以2.5μm为例,使用自来水配制250μm的草甘膦自来水溶液,取6μl 250μm的草甘膦自来水溶液与20μl含ce
3+
溶液(0.5mm)和4μl焦磷酸钠溶液(1mm)混合,然后将混合物在室温下以600rpm振荡5min。在300nm激发下记录346nm处的荧光光谱和荧光强度,将得到的荧光强度代入实施例3得到的线性方程中,得到自来水中的草甘膦的浓度。
64.表1实验结果(回收率)
[0065][0066]
从表1中可以得出,基于测定后的荧光强度,代入线性方程后经计算得到对应添加样品浓度为0.44和2.17μm,即测定浓度,经计算回收率在标准范围(70-110%)内,证明该方法可用于自来水中草甘膦的检测。
[0067]
实施例5
[0068]
检测时间的确定:
[0069]
具体实验步骤:第一部分:固定与草甘膦作用15min,即在ce
3+
与ppi结合0、5、10、15、20min后再与草甘膦混合15min,计算荧光相对变化率,确定最佳结合时间为5min。第二部分:与第一部分同样,固定最佳结合时间5min,即在ce
3+
与ppi结合5min后,加入草甘膦与其混合0、5、10、15、20min,计算荧光相对变化率,确定最佳作用时间为0min。
[0070]
由上述可知,检测过程分为两部分,第一部分是ce
3+
与ppi结合生成ce-ppi,第二部分是草甘膦与ce-ppi作用生成三元混合物。通过实验发现,第一部分过程经过5分钟时荧光响应值即可基本稳定(结果见图9),第二部分过程直接将草甘膦与ce
3+
、ppi一起混合经过第一部分过程荧光响应值即可基本稳定(结果见图10),故整个过程检测时间为5min。
[0071]
对比例
[0072]
按照以下参考文献来检测草甘膦的浓度,具体结果见表2,参考文献如下:
[0073]
[1]项阳.草甘膦及氨甲基磷酸残留量检测方法综述[j].吉林公安高等专科学校学报,2011,26(02):68-71.
[0074]
[2]郑亚丽,冯小康,朱强.高效液相色谱法测定土壤中草甘膦的残留[j].化工时
刊,2021,35(07):12-15+59.
[0075]
[3]张云峰,赵森,常靖,任昕昕,王爱华,赵鹏,董林沛,吴小军,张景然,刘冰洁.非衍生化高效液相色谱-串联质谱法快速检测生物体液中草甘膦、草铵膦及代谢物[j].分析测试学报,2021,40(04):571-576.
[0076]
[4]de almeida,l.k.s.;chigome,s.;torto,n.;frost,c.l.;pletschke,b.i.,a novel colorimetric sensor strip for the detection of glyphosate in water.sensors and actuators b-chemical 2015,206,357-363。
[0077]
表2检测限结果
[0078]
分析方法参考文献检出限(μgl-1
)荧光法19.8比色法2100电化学方法310荧光法45荧光法本发明2.37(0.014μm)
[0079]
从表2中可以得出,经对比可得,本发明的检出限优于绝大多数现有方法。
[0080]
以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1