一种异噁唑啉类兽药中间体铵盐有关杂质的分析方法与流程
1.本发明属于药物中间体分析领域,具体涉及一种异噁唑啉类兽药中间体铵盐有关杂质的分析方法。
背景技术:
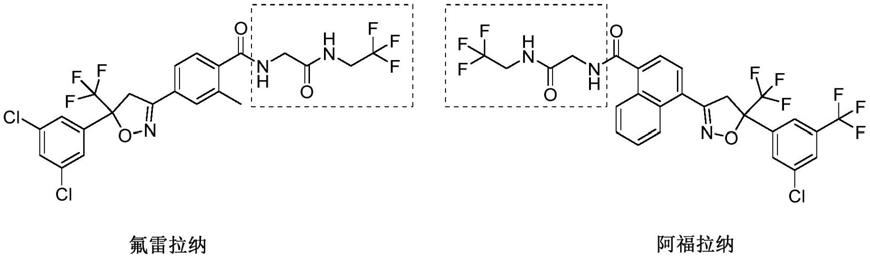
2.氟雷拉纳(fluralaner)和阿福拉纳(afoxolaner)属于高端长效抗寄生虫药物,其通过干扰寄生虫的γ-氨基丁酸(gaba)门控氯离子通道发挥作用导致神经系统过度兴奋而死亡。fluralaner是第一个提供长达12周的针对跳蚤和蜱虫的口服咀嚼杀体外寄生虫药。一粒咀嚼片或一剂局部用溶液可提供广谱和持久的保护,在2小时内开始杀死跳蚤,并控制4种蜱虫(黑腿蜱,美洲犬蜱,棕色犬蜱和孤星蜱)。afoxolaner作为国内首个同时兼杀蜱虫和跳蚤的口服驱虫药,口服后可快速起效,通过阿福拉纳分子作用于节肢动物的神经传递突触,迅速使跳蚤和蜱虫在极度兴奋之下导致死亡,服用一片效果可持30天。
3.氟雷拉纳与阿福拉纳化学结构类似,它们拥有相同的一个关键片段2-氨基-n-(2,2,2-三氟乙基)乙酰胺,氟雷拉纳与阿福拉纳其化学结构如下:
[0004][0005]
2-氨基-n-(2,2,2-三氟乙基)乙酰胺作为构成氟雷拉纳和阿福拉纳重要片段,目前其报道的合成路线主要的合成路线如下几种:
[0006][0007]
由路线以及结构可知,主要由两部分构成(如下),由此可知对于甘氨酸和三氟乙胺盐这两个主要杂质的研究对于2-氨基-n-(2,2,2-三氟乙基)乙酰胺起着至关重要的作用。
[0008][0009]
为了更好地控制2-氨基-n-(2,2,2-三氟乙基)乙酰胺以及阿福拉纳和氟雷拉纳的质量,本发明对自制以及市面上大部分的2-氨基-n-(2,2,2-三氟乙基)乙酰胺中的有关物质进行了系统的研究,发现主要的杂质均为甘氨酸和三氟乙胺盐,目前关于2-氨基-n-(2,2,2-三氟乙基)乙酰胺的质量研究的报道并不多。
技术实现要素:
[0010]
本发明的目的在于克服现有技术的至少一个不足,提供一种方便对2-氨基-n-(2,2,2-三氟乙基)乙酰胺进行质量研究的通用办法。
[0011]
本发明所采取的技术方案是:
[0012]
一种异噁唑啉类兽药中间体铵盐有关杂质的分析方法,所述中间体为2-氨基-n-(2,2,2-三氟乙基)乙酰胺,所述有关杂质为三氟乙胺盐酸盐以及甘氨酸,包括:
[0013]
使用稀释剂溶解所述中间体铵盐和对照品,得试样溶液和对照品溶液;
[0014]
使用反相高效液相色谱仪检测甘氨酸和使用气相色谱检测三氟乙胺盐酸盐:
[0015]
使用反相高效液相色谱仪检测甘氨酸包括:
[0016]
使用反相高效液相色谱仪检测试样溶液中中间体铵盐及其杂质,确定色谱图,所述反相高效液相色谱的条件为:流动相:a:乙腈;b:七氟丁酸溶液;洗脱方式:梯度洗脱,以体积分数计,所述梯度洗脱具体为0min~5.0min,10%~20%a,90~80%b;5min~15min,20%~40%a,80~60%b 15min~20min,40%~65%a,60%~35%b;20min~22min,65%~80%a,35~20%b,20min~27min,80%~10%a,20%~90%b;40%~65%a,27min~35min,10%a,90%b;所述反相高效液相色谱检测器为电雾式检测器;
[0017]
根据液相色谱结果,确定所述中间体和甘氨酸的量;
[0018]
使用气相色谱检测三氟乙胺盐酸盐,所述气相色谱的条件为:样口温度为200~240℃,检测器的温度为240~280℃,载气为氮气,分流比为(10~20):1。
[0019]
在一些实例中,所述反相高效液相色谱中流动相b中七氟丁酸溶液的体积浓度为0.1%~0.15%。
[0020]
在一些实例中,所述反相高效液相色谱中流动相的流速为0.8~1.0ml/min。
[0021]
在一些实例中,所述反相高效液相色谱的色谱柱温为30℃~40℃。
[0022]
在一些实例中,所述反相高效液相色谱的色谱柱为venusil xbp c18(l)色谱柱:5μm,4.6x250mm。
[0023]
在一些实例中,所述电雾式检测器cad参数为:载气为氮气,压力35psi,雾化温度35℃。
[0024]
在一些实例中,所述气相色谱的升温程序为:起始温度30~50℃恒温3min,8~12℃/min的速率升温至180℃~240℃,恒温3~8min。
[0025]
在一些实例中,配制对照品溶液和供试品溶液的稀释剂为0.2%碳酸钠溶液。
[0026]
在一些实例中,所述气相色谱的色谱柱为安捷伦30m*0.32mm,1.8μm。
[0027]
在一些实例中,所述反相高效液相色谱的进样量为10~20μl。
[0028]
在一些实例中,所述气相色谱的进样量为0.5~1.5ml。
[0029]
本发明的有益效果是:
[0030]
本发明的方法能够有效分离2-氨基-n-(2,2,2-三氟乙基)乙酰胺的杂质,该方法高效、简便、灵敏度高、分离效果好。
附图说明
[0031]
图1是实施例1高效液相方法供试品溶液图谱;
[0032]
图2是实施例2高效液相方法空白溶液图谱;
[0033]
图3是实施例2高效液相方法系统适用性溶液图谱;
[0034]
图4是实施例8气相方法系统供试品溶液图谱;
[0035]
图5是实施例9气相方法空白溶液图谱;
[0036]
图6是实施例9气相方法系统适用性溶液图谱;
[0037]
图7是实施例11气相色谱的线性回归回归曲线。
具体实施方式
[0038]
下面进一步列举实施例以详细说明本发明。同样应理解,以下实施例只用于对本发明进行进一步说明,不能理解为对本发明保护范围的限制,本领域技术人员根据本发明阐述的原理做出的一些非本质的改进和调整均属于本发明的保护范围。下述示例具体的工艺参数等也仅是合适范围中的一个示例,即本领域技术人员可以通过本文的说明做合适范围内的选择,而并非要限定于下文示例的具体数据。
[0039]
下述实施例中所称的杂质c为三氟乙胺盐酸盐,杂质d为甘氨酸。
[0040]
方便比较起见,以下实施例中,标准对照品溶液、异噁唑啉类杀虫剂参比溶液、试样溶液的配制与反相高效液相色谱检测步骤相同,区别在于反相高效液相色谱(或气相色谱)检测条件不同,则标准对照品溶液、异噁唑啉类杀虫剂参比溶液、试样溶液的配制与反相高效液相色谱检(或气相色谱)测步骤如下:
[0041]
对于杂质d的高效液相检测方法:
[0042]
实施例1:
[0043]
1)标准对照品溶液的配制:准确称取2-氨基-n-(2,2,2-三氟乙基)乙酰胺标准品(以下简称ay标准品加入稀释剂(稀释剂为每1ml中含20μg抗坏血酸溶液,下同)溶解,配制成每毫升含ay标准品每1ml中含15mg工作标的对照品溶液。
[0044]
2)供试品溶液的配制:准确称取2-氨基-n-(2,2,2-三氟乙基)乙酰胺供试品,加稀释剂溶解制成每1ml中含供试品75μg的溶液。
[0045]
3)杂质d贮备液:精密称定杂质d对照品10mg,置10ml容量瓶中,加稀释剂溶解并定容至刻度,制成每1ml中各含1mg杂质d的贮备液。
[0046]
4)仪器:高效液相色谱仪(cad检测器);
[0047]
色谱柱:venusil xbp c18(l),5μm,4.6x250mm;
[0048]
流动相a:乙腈;
[0049]
流动相b:0.1%七氟丁酸;
[0050]
流速:1ml/min;
[0051]
柱温:35℃;
[0052]
cad参数:载气为氮气,压力35psi,雾化温度35℃;
[0053]
进样量:20μl;
[0054]
洗脱条件如表1所示。
[0055]
表1、色谱洗脱条件
[0056][0057]
图1是实施例1高效液相方法供试品溶液图谱,从图中可以看出,杂质可以有效分离。
[0058]
实施例2:专属性
[0059]
按照实施例1配制对照品溶液和供试品溶液以及杂质d贮备液;
[0060]
系统适用性溶液的配制:配制每1ml中含15mg供试品及杂质a、杂质b、杂质d各75μg的系统适用性溶液;
[0061]
取供试品溶液配制各种破坏溶液:
[0062]
光照破坏溶液:取供试品贮备液至透明ep管中放置254nm紫外光下照射24小时;
[0063]
氧化破坏溶液:取供试品贮备液加3%h2o2,室温放置4小时;
[0064]
高温破坏溶液:取供试品贮备液1ml置透明ep管,在60℃水浴中放置3小时后冷却;
[0065]
酸破坏溶液:取供试品贮备液加0.1mol/l hcl,室温放置4小时,加0.1mol/l naoh中和;
[0066]
碱破坏溶液:取供试品贮备液,加0.1mol/l naoh室温放置4小时,加0.1mol/l hcl中和。
[0067]
对不同破坏溶液进行检测,结果如下表2、表3所示。
[0068]
表2、xs-ay各杂质定位结果
[0069][0070]
注:杂质定位结果来自系统适用性溶液。
[0071]
表3、专属性破坏实验结果
[0072][0073]
结论:空白溶剂无干扰(图2),系统适用性溶液中xs-ay与杂质d分离度为6.81,大于1.5,杂质d与前后相邻杂质峰的分离度分别为na、9.16,大于1.5,符合要求(图3)。杂质限度溶液与系统适用性溶液保留时间基本一致;破坏实验中回收率为93.77%-101.34%,符合要求。破坏后的供试品溶液无与主峰保留时间相同或相近的色谱峰。
[0074]
实施例3:检测限及定量限
[0075]
1)杂质d贮备液:精密称定杂质d对照品10mg,置10ml容量瓶中,加稀释剂溶解并定容至刻度,制成每1ml中各含1mg杂质d的贮备液。
[0076]
根据实施例2中各有关物质限度溶液色谱图可初步得出各组分的信噪比,取贮备液进行逐步稀释,找出信噪比为2~4对应的浓度,定为检测限浓度;信噪比为8~12对应的浓度,定为定量限浓度。结果如下表4,表5所示。
[0077]
表4、杂质d对照品检测限结果
[0078][0079]
表5、杂质d对照品定量限结果
[0080][0081]
结论:3份检测限溶液信噪比均大于3,保留时间rsd为0.09%,不大于1.0%,峰面
积rsd为4.52%,不大于10.0%,符合要求。6份定量限溶液信噪比均大于10,保留时间rsd为0.04%,不大于1.0%,峰面积rsd为3.62%,不大于10.0%,符合要求。故:最终确定有关物质的检测限2.65μg/ml,定量限7.96μg/ml。试验结果表明,本发明的的检测方法对各有关物质均具备较高的灵敏度。
[0082]
实施例4:线性关系
[0083]
s1:杂质d贮备液,加稀释剂分别稀释成供试品浓度的0.05%、0.10%、0.30%、0.50%、1.00%的溶液(限度0.5%)。每份样品检测3次;
[0084]
s2:按照实施例1中的高效液相色谱条件测定峰面积,以浓度为横坐标,峰面积为纵坐标,得到各有关物质的线性回归方程,结果见表6。
[0085]
表6、校正因子结果
[0086][0087]
结论:xs-ay线性方程为y=0.0692x+0.1366,r=0.9973,大于0.990,截距为2.44%,符合要求;杂质d线性方程为y=0.0497x+0.0263,r=0.9995,大于0.990,截距为0.60%,符合要求;校正因子为1.39。
[0088]
实施例5:准确度试验
[0089]
s1:按实施例1配制对照品溶液
[0090]
准确度基础溶液:称取供试品加稀释剂制成含供试品15mg/ml的溶液
[0091]
准确度1加标溶液:称取供试品和杂质d贮备液用稀释剂稀释配制成加进去的杂质对照品浓度为杂质限度50%溶液平行配制3份,依次命名为50%-1、50%-2、50%-3。
[0092]
准确度2加标溶液:称取供试品和杂质d贮备液用稀释剂稀释配制成加进去的杂质对照品浓度为杂质限度100%溶液平行配制3份,依次命名为100%-1、100%-2、100%-3。
[0093]
准确度3加标溶液:称取供试品和杂质d贮备液用稀释剂稀释配制成加进去的杂质对照品浓度为杂质限度150%溶液平行配制3份,依次命名为150%-1、150%-2、150%-3。
[0094]
s2:按照实施例1中的高效液相色谱条件测定样品溶液的峰面积,计算加杂供试品溶液中有关物质的回收率,结果如下表7。
[0095]
表7、准确度结果
[0096][0097]
结论:基础溶液中杂质d未检出,不同浓度加标溶液的回收率为94.43%~97.85%,平均回收率为95.97%,在90.0%~108.0%范围内,9份回收率rsd为1.34%,小于10.0%,符合要求。
[0098]
实施例6:耐用性
[0099]
s1:按照实施例1配制1份对照品溶液和2份供试品溶液;s2:按照实施例1中的高效液相色谱条件,分别在柱温=30
±
5℃、流速=1.2
±
0.3ml/min条件下检测样品溶液,计算在不同柱温、流速下供试品溶液中有关物质含量,结果如表8、表9所示。
[0100]
表8、不同柱温耐用性结果
[0101][0102]
表9、不同流速耐用性结果
[0103][0104]
试验结果表明,柱温波动不超过5℃以及流速变化不超过0.3ml/min情况下对2-氨基-n-(2,2,2-三氟乙基)乙酰胺供试品溶液中有关物质的影响均在可接受范围
[0105]
实施例7:稳定性
[0106]
s1:按照实施例1配制对照品溶液和供试品溶液;
[0107]
s2:按照实施例1中的高效液相色谱条件测定样品溶液,分别在配制后的0、3、6、9、12、15、18、21、24小时进行测定。计算得到供试品溶液中有关物质含量结果如表10所示。
[0108]
表10、供试品加标溶液常温稳定性结果
[0109][0110]
结论:对照品溶液常温放置24小时,主峰面积rsd为1.54%,小于5.0%,供试品加标溶液常温放置24小时,杂质d峰面积rsd为1.56%,小于5.0%,符合要求。对照品溶液和供试品加标溶液常温放置24小时稳定。
[0111]
对于杂质c的气相检测方法:
[0112]
实施例8
[0113]
1)溶剂(0.2%碳酸钠溶液):称取0.5g无水碳酸钠于250ml容量瓶中,加纯化水溶解并稀释至刻度即可;
[0114]
2)空白溶液:0.2%碳酸钠溶液;
[0115]
3)对照溶液:精密称取杂质c1mg于10ml容量瓶中,并用0.2%碳酸钠溶液溶解并稀释至刻度,作为对照溶液贮备液;取其1ml对照溶液贮备液于10ml容量瓶中,并用0.2%碳酸钠溶液稀释至刻度即可,取1ml该溶液于顶空瓶中(亦作为系统适用性溶液);
[0116]
4)供试溶液:精密称取样品0.1g于10ml容量瓶中,并用0.2%碳酸钠溶液溶解并稀释至刻度,取其2ml上述溶液与10ml容量瓶中,并用0.2%碳酸钠溶液稀释至刻度即可,取1ml该溶液于顶空瓶中。
[0117]
5)仪器:气相色谱仪(fid检测器);
[0118]
色谱柱:db-624(安捷伦30m*0.32mm,1.8μm);
[0119]
载气:氮气;
[0120]
柱温:50℃;
[0121]
分流比:15:1;
[0122]
流速:2.0ml/min;
[0123]
运行时间:15min;
[0124]
孵化温度:70℃;
[0125]
环/样品管路温度:80℃;
[0126]
平衡时间:30min;
[0127]
检测器温度为250℃;
[0128]
进样口温度为220℃;
[0129]
进样量:1ml。
[0130]
图4是实施例8气相方法系统供试品溶液图谱。从图中可以看出,杂质可以有效分离,利于检测。
[0131]
实施例9:系统适用性及专属性
[0132]
按照实施例8配制对照溶液(系统适用性溶液)和供试品溶液
[0133]
表11系统适用性溶液结果
[0134][0135]
表12专属性定位结果
[0136][0137]
注释
①
:空白溶液没有干扰
[0138]
结果和结论:
[0139]
1)空白溶液没有干扰(图5),供试溶液中杂质c的保留时间与对照溶液中一致,符合标准;
[0140]
2)连续5针系统适用性溶液杂质c峰面积rsd为0.78%(图6),符合要求。
[0141]
实施例10:检测限及定量限
[0142]
1)杂质c贮备液:精密称定杂质c对照品10mg,置10ml容量瓶中,加溶剂(0.2%碳酸钠溶液)溶解并定容至刻度,制成每1ml中各含1mg杂质c的贮备液。
[0143]
根据实施例2中各有关物质限度溶液色谱图可初步得出各组分的信噪比,取贮备液进行逐步稀释,找出信噪比为2~4对应的浓度,定为检测限浓度;信噪比为8~12对应的浓度,定为定量限浓度,如下表13,表14所示。
[0144]
表13、杂质c定量限结果
[0145][0146]
表14杂质c检测限结果
[0147]
名称浓度(μg/ml)s/n检测限溶液0.11967.0
[0148]
结论:杂质c检测限浓度为0.1196μg/ml,s/n为7.0;杂质c定量限浓度为0.2392μg/ml,最小s/n为14.3,峰面积rsd为4.8%,符合标准。
[0149]
实施例11:线性关系试验
[0150]
s1:杂质c贮备液,加0.2%碳酸钠溶液分别稀释成供试品浓度的0.01%、0.05%、0.05%、0.10%、0.50%、0.75%的溶液(限度0.5%)。每份样品检测3次;
[0151]
s2:按照实施例1中的气相色谱条件测定峰面积,以浓度为横坐标,峰面积为纵坐标,得到各有关物质的线性回归方程,结果见表15,图7所示。
[0152]
表15、线性结果
[0153]
序号浓度(μg/ml)峰面积(pa*min)限度浓度的百分比线性10.21390.00802.0%线性21.06960.039810.0%线性35.34790.200050.0%线性410.69580.3648100.0%线性516.04360.5558150.0%
[0154]
结论:杂质c在0.2139μg/ml-16.0436μg/ml浓度范围内的线性关系良好,回归方程为y=0.0343x+0.0048,r值为0.9995,符合标准。
[0155]
实施例12:准确度
[0156]
s1:按实施例8配制对照品溶
[0157]
准确度基础溶液:称取供试品加稀释剂制成含供试品15mg/ml的溶液
[0158]
准确度1加标溶液:称取供试品和杂质c贮备液用稀释剂稀释配制成加进去的杂质对照品浓度为杂质限度50%溶液平行配制3份,依次命名为50%-1、50%-2、50%-3。
[0159]
准确度2加标溶液:称取供试品和杂质c贮备液用稀释剂稀释配制成加进去的杂质对照品浓度为杂质限度100%溶液平行配制3份,依次命名为100%-1、100%-2、100%-3。
[0160]
准确度3加标溶液:称取供试品和杂质c贮备液用稀释剂稀释配制成加进去的杂质对照品浓度为杂质限度150%溶液平行配制3份,依次命名为150%-1、150%-2、150%-3。
[0161]
s2:按照实施例8中的气相色谱条件测定样品溶液的峰面积,计算加杂供试品溶液中有关物质的回收率,结果如下表16。
[0162]
表16、xs-ay杂质c准确度结果
[0163][0164]
结论:xs-ay杂质c的回收率在94.69%~109.93%之间,平均回收率为99.19%,rsd为4.3%,符合标准。
[0165]
实施例13:耐用性
[0166]
s1:按照实施例8配制基础溶液和标准溶液以及甲标溶液;
[0167]
s2:按照实施例8中的气相色谱条件,分别在柱温=70
±
5℃、流速=2.0
±
0.5ml/min条件下检测样品溶液,计算在不同柱温、流速下供试品溶液中有关物质含量,结果如表17、表18、表19、表20、表21、表22所示。
[0168]
表17色谱条件变动参数
[0169]
色谱参数规定值变动范围流速(ml/min)2.01.5和2.5顶空平衡温度(℃)7065和75
[0170]
表18、流速为1.5ml/min的回收率结果
[0171][0172]
表19、流速为2.5ml/min的回收率结果
[0173][0174]
表20、顶空平衡温度为65℃的回收率结果
[0175][0176]
表21、顶空平衡温度为75℃的回收率结果
[0177][0178]
表22、不同测定条件参数的结果对比
[0179][0180]
注释
①
:该回收率采用准确度项目时的平均回收率。
[0181]
结论:在顶空平衡温度
±
5℃,流速变化
±
0.5ml/min时,回收率在90.93%~99.19%之间,符合要求。
[0182]
实施例14:稳定性
[0183]
s1:按照实施例8配制对照品溶液和供试品溶液各三份;
[0184]
s2:按照实施例8中的气相色谱条件测定样品溶液,分别在配制后的0、6、
[0185]
12小时进行测定。计算得到供试品溶液中有关物质含量结果如表23、表24所示。
[0186]
表23、供试溶液稳定性结果
[0187][0188]
表24、对照溶液稳定性结果
[0189][0190]
(4)结论:
[0191]
1、供试溶液在0h、6h和12h测得杂质c峰面积的rsd为8.0%,符合标准;
[0192]
2、对照溶液在0h、6h和12h测得杂质c峰面积的rsd为7.2%,符合标准。
[0193]
以上是对本发明所作的进一步详细说明,不可视为对本发明的具体实施的局限。对于本发明所属技术领域的普通技术人员来说,在不脱离本发明构思的简单推演或替换,都在本发明的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1