一种检测低毒类农药含量的方法
1.本发明涉及分析检测技术领域,具体涉及一种检测低毒类农药含量的方法。
背景技术:
2.2019年全球作物用农药市场的销售额为598.27亿美元,新型低毒农药如甲氧基丙烯酸酯类杀菌剂、三唑类杀菌剂、新烟碱类杀虫剂以及酰胺类除草剂,在全球的市场地位也处于领先优势。此类农药在农作物上的半衰期短,施药后易进入土壤,不易降解,通过地表径流和渗透作用会对水生生物的繁殖能力造成影响。如双氟磺草胺具有较强的亲水性,更容易进入水体,成为水体污染的原因之一。
3.水体中多残留农药的传统前处理方法为液液萃取法和固相萃取法,液液萃取法往往需要耗费大量有机溶剂,有机溶剂的选择难以兼容多种类农药分析,无法凸显绿色分析化学的优势;固相萃取法通常包含较为复杂的富集程序,成本较高。
技术实现要素:
4.本发明的目的在于一种检测低毒类农药含量的方法,本发明基于h-beta型分子筛建立了分散固相萃取结合高效液相色谱-串联质谱检测低毒类农药含量的方法,操作简单,且待测农药定性定量准确度高,灵敏度高。
5.为了实现上述发明目的,本发明提供以下技术方案:
6.本发明提供了一种检测低毒类农药含量的方法,包括以下步骤:
7.采用h-beta型分子筛对待测样品进行涡旋吸附,得到吸附样品;
8.采用解吸附液将所述吸附样品进行涡旋超声解吸附,得到待测液;所述解吸附液包括乙腈和甲醇;
9.采用高效液相色谱-串联质谱检测所述待测液中低毒类农药的含量;
10.所述低毒类农药包括乙草胺、苯并烯氟菌唑、环氟菌胺、吡氟酰草胺、环酰菌胺、氟啶虫酰胺、双氟磺草胺、唑嘧磺草胺、氟吡菌胺、氟吡菌酰胺、吡唑萘菌胺、双炔酰菌胺、氟唑菌苯胺、吡噻菌胺、丙草胺、毒草胺、敌稗、氟胺磺隆、啶酰菌胺、呋虫胺、稻瘟酰胺、氟苯虫酰胺、氟噻草胺、氟酰胺、氟唑菌酰胺、吡虫啉、氯噻啉、噻虫嗪、烯肟菌胺、啶氧菌酯和肟菌酯中的至少一种。
11.优选地,所述h-beta型分子筛的硅铝摩尔比为20~40,粒度为50~100μm。
12.优选地,所述h-beta型分子筛和待测样品的用量比为50mg:(20~90)ml。
13.优选地,所述涡旋吸附的温度为5~50℃,转速为500~2000rpm,时间为5~30min。
14.优选地,所述解吸附液中乙腈与甲醇的体积比为(1~3):1。
15.优选地,所述解吸附液中还包括甲酸,所述解吸附液中甲酸的体积分数≤0.5%。
16.优选地,所述h-beta型分子筛与解吸附液的用量比为50mg:(1~2)ml。
17.优选地,所述涡旋超声解吸附包括依次进行的涡旋解吸附和超声解吸附;所述涡旋解吸附的温度为10~40℃,转速为500~2000rpm,时间为1~10min;所述超声解吸附的温
度为10~40℃,时间为1~10min。
18.优选地,所述高效液相色谱-串联质谱中高效液相色谱的检测条件包括:流动相体系为流动相a和流动相b,所述流动相a为甲醇,所述流动相b为乙酸铵水溶液,所述乙酸铵水溶液的浓度为2~10mmol/l;所述流动相体系的流速为0.2~0.4ml/min;洗脱方式为梯度洗脱,所述梯度洗脱的程序为:
19.0.0~0.5min,流动相a的体积分数为2%;0.5~15.0min,流动相a的体积分数由2%增加至98%;15.0~17.0min,流动相a的体积分数维持在98%;17.0~17.1min,流动相a的体积分数由98%降低至2%;17.1~20.0min,流动相a的体积分数维持在2%。
20.优选地,所述高效液相色谱-串联质谱中质谱的检测条件包括:离子源为电喷雾电离源;检测方式为正离子模式;离子化电压为4000~5500v;离子源温度为400~550℃;气帘气压强为20~30psi,喷雾气压强为40~50psi;辅助加热气压强为40~50psi;碰撞气压强为4~7psi。
21.本发明提供了一种检测低毒类农药含量的方法。本发明基于h-beta型分子筛建立了分散固相萃取结合高效液相色谱-串联质谱检测低毒类农药含量的方法,采用h-beta型分子筛作为吸附材料能够实现待测农药的高效吸附,采用包括乙腈和甲醇的溶液作为解吸附液能够实现待测农药的有效解吸附,最后采用高效液相色谱-串联质谱进行检测。本发明提供的方法操作简单,且待测农药定性定量准确度高,灵敏度高。实施例的结果显示,本发明提供的方法具有较高的准确度(平均回收率》62.2%)、精密度(相对标准偏差《18.0%)和灵敏度(水的检出限0.04~0.1ng/ml,定量限为0.08~0.2ng/ml),满足了水中农药检测的要求。
附图说明
22.图1为本发明实施例中对实际水样品中农药进行检测的流程图。
具体实施方式
23.本发明提供了一种检测低毒类农药含量的方法,包括以下步骤:
24.采用h-beta型分子筛对待测样品进行涡旋吸附,得到吸附样品;
25.采用解吸附液将所述吸附样品进行涡旋超声解吸附,得到待测液;所述解吸附液包括乙腈和甲醇;
26.采用高效液相色谱-串联质谱检测所述待测液中低毒类农药的含量;
27.所述低毒类农药包括乙草胺、苯并烯氟菌唑、环氟菌胺、吡氟酰草胺、环酰菌胺、氟啶虫酰胺、双氟磺草胺、唑嘧磺草胺、氟吡菌胺、氟吡菌酰胺、吡唑萘菌胺、双炔酰菌胺、氟唑菌苯胺、吡噻菌胺、丙草胺、毒草胺、敌稗、氟胺磺隆、啶酰菌胺、呋虫胺、稻瘟酰胺、氟苯虫酰胺、氟噻草胺、氟酰胺、氟唑菌酰胺、吡虫啉、氯噻啉、噻虫嗪、烯肟菌胺、啶氧菌酯和肟菌酯中的至少一种,优选为乙草胺、苯并烯氟菌唑、环氟菌胺、吡氟酰草胺、环酰菌胺、氟啶虫酰胺、双氟磺草胺、唑嘧磺草胺、氟吡菌胺、氟吡菌酰胺、吡唑萘菌胺、双炔酰菌胺、氟唑菌苯胺、吡噻菌胺、丙草胺、毒草胺、敌稗、氟胺磺隆、啶酰菌胺、呋虫胺、稻瘟酰胺、氟苯虫酰胺、氟噻草胺、氟酰胺、氟唑菌酰胺、吡虫啉、氯噻啉、噻虫嗪、烯肟菌胺、啶氧菌酯和肟菌酯。
28.本发明基于h-beta型分子筛建立了分散固相萃取结合高效液相色谱-串联质谱检
测低毒类农药的方法,操作简单,基于所述h-beta型分子筛的表面多孔性质,能够实现低毒类农药的有效吸附,且待测农药定性定量准确度高,灵敏度高。下面首先对本发明所述h-beta型分子筛进行详细说明。
29.在本发明中,所述h-beta型分子筛的硅铝摩尔比优选为20~40,更优选为26,粒度优选为50~100μm。在本发明中,所述h-beta型分子筛的制备方法,优选包括以下步骤:
30.将氢氧化钠、铝源、四乙基氢氧化胺、硅源和水混合,将得到的凝胶进行水热反应后第一煅烧,得到na-beta型分子筛;
31.将所述na-beta型分子筛与铵盐水溶液混合,进行离子交换后第二煅烧,得到h-beta型分子筛。
32.在本发明中,若没有特殊说明,所采用的试剂均为本领域技术人员所熟知的市售商品。
33.本发明将氢氧化钠、铝源、四乙基氢氧化胺、硅源和水混合,将得到的凝胶进行水热反应后第一煅烧,得到na-beta型分子筛。在本发明中,所述铝源优选包括naalo2和/或al2(so4)3,更优选为naalo2。在本发明中,所述硅源优选包括硅胶粉、白炭黑和粗孔硅胶中的一种或几种,更优选为硅胶粉。在本发明中,所述铝源以al2o3计,所述硅源以sio2计,氢氧化钠以na2o计,所述凝胶中al2o3和na2o的摩尔比优选为1:(2~10),更优选为1:(3~5);所述al2o3与四乙基氢氧化胺的摩尔比优选为1:(5~15),更优选为1:(6~9);所述al2o3与sio2的摩尔比优选为1:(20~40),更优选为1:26;所述al2o3与h2o的摩尔比优选为1:(12~50),更优选为1:(16~20)。在本发明中,所述氢氧化钠、铝源、四乙基氢氧化胺、硅源和水的混合优选为:将氢氧化钠、铝源和四乙基氢氧化胺溶解于水中,在搅拌条件下加入硅源后继续搅拌;本发明对所述搅拌的速度没有特殊限定,能够将原料混合均匀即可;所述继续搅拌的时间优选为2~6h,更优选为2h。
34.在本发明中,所述水热反应的温度优选为140~160℃,更优选为145℃;所述水热反应的时间优选为36~60h,更优选为48h。所述水热反应后,本发明优选还包括将所述水热反应得到的反应液冷却至室温后固液分离,将得到的固体产物水洗后干燥,得到na-beta型分子筛前驱体,然后将所述na-beta型分子筛前驱体进行第一煅烧,得到na-beta型分子筛。本发明对所述冷却的方式没有特殊限定,采用本领域技术人员熟知的冷却方式即可,具体如自然冷却。本发明对所述固液分离的方式没有特殊限定,采用本领域技术人员熟知的固液分离方式即可,具体如过滤或抽滤。在本发明中,所述水洗用水优选包括去离子水和/或蒸馏水。在本发明中,所述干燥的温度优选为80~120℃,更优选为100℃;所述干燥的时间优选为10~14h,更优选为12h。在本发明中,所述第一煅烧的温度优选为500~600℃,更优选为550℃;所述第一煅烧的时间优选为5~8h,更优选为6~7h;所述第一煅烧优选在空气氛围中进行。在本发明中,所述第一煅烧的目的是除去四乙基氢氧化胺(模板剂)。
35.得到na-beta型分子筛后,本发明将所述na-beta型分子筛与铵盐水溶液混合,进行离子交换后第二煅烧,得到h-beta型分子筛。在本发明中,所述铵盐水溶液优选为nh4cl水溶液,所述铵盐水溶液的浓度优选为0.5~2mol/l,更优选为1mol/l。在本发明中,所述beta型分子筛的质量与铵盐水溶液的体积之比(固液比)优选为1g:(20~90)ml,更优选为1g:50ml。本发明对所述na-beta型分子筛与铵盐水溶液混合的方式没有特殊限定,采用本领域技术人员熟知的混合方式能够将原料混合均匀即可,具体如搅拌混合。
36.在本发明中,所述离子交换反应的温度优选为80~90℃,更优选为85℃;所述离子交换反应的时间优选为1~3h,更优选为2h;所述离子交换反应过程中铵根离子将na-beta型分子筛中的钠离子交换出来。在本发明中,所述第二煅烧的温度优选为500~600℃,更优选为550℃;所述第二煅烧的时间优选为6~9h,更优选为6~7h;所述第二煅烧优选在空气氛围中进行。在本发明中,所述第二煅烧的目的是将铵根离子分解,从而获得h-beta型分子筛。
37.得到h-beta型分子筛后,本发明采用h-beta型分子筛对待测样品进行涡旋吸附,得到吸附样品。在本发明中,所述待测样品优选包括水,所述水优选包括城市河流水或稻田水。在本发明中,所述h-beta型分子筛和待测样品的用量比优选为50mg:(20~90)ml,更优选为50mg:50ml。在本发明中,所述涡旋吸附的温度优选为5~50℃,更优选为20~30℃,具体可以为室温(25℃);所述涡旋吸附的转速优选为500~2000rpm,更优选为1000~1500rpm;所述涡旋吸附的时间优选为5~30min,更优选为15~20min。所述涡旋吸附后,本发明优选还包括将所述涡旋吸附得到的吸附料液进行离心分离,弃去全部上清液,得到的固体组分为吸附样品。在本发明中,所述离心分离的温度优选为室温;所述离心分离的速度优选为1000~6000rpm,更优选为5000rpm;所述离心分离的时间优选为1~9min,更优选为5min。
38.得到吸附样品后,本发明采用解吸附液将所述吸附样品进行涡旋超声解吸附,得到待测液;所述解吸附液包括乙腈和甲醇。在本发明中,所述解吸附液中乙腈与甲醇的体积比优选为(1~3):1,更优选为3:1。在本发明中,所述解吸附液中优选还包括甲酸,所述解吸附液中甲酸的体积分数优选≤0.5%,更优选为0.4~0.5%。在本发明中,所述h-beta型分子筛与解吸附液的用量比优选为50mg:(1~2)ml,更优选为50mg:2ml。
39.在本发明中,所述涡旋超声解吸附优选包括依次进行的涡旋解吸附和超声解吸附;所述涡旋解吸附的温度优选为10~40℃,更优选为20~30℃,具体可以为室温;所述涡旋解吸附的转速优选为500~2000r/min,更优选为2000r/min;所述涡旋解吸附的时间优选为1~10min,更优选为3~5min;所述超声解吸附的温度优选为10~40℃,更优选为20~30℃,具体可以为室温;所述超声解吸附的时间优选为1~10min,更优选为5min。所述涡旋超声解吸附后,本发明优选还包括将所述涡旋超声解吸附得到的解吸附料液进行离心分离,将得到的上清液进行微膜过滤,得到待测液。在本发明中,所述离心分离的温度优选为室温;所述离心分离的速度优选为1000~6000rpm,更优选为3000~5000rpm;所述离心分离的时间优选为1~5min,更优选为3~4min。在本发明中,所述微膜过滤优选采用0.22μm尼龙过滤器进行。
40.得到待测液后,本发明采用高效液相色谱-串联质谱检测所述待测液中低毒类农药的含量;所述低毒类农药包括乙草胺、苯并烯氟菌唑、环氟菌胺、吡氟酰草胺、环酰菌胺、氟啶虫酰胺、双氟磺草胺、唑嘧磺草胺、氟吡菌胺、氟吡菌酰胺、吡唑萘菌胺、双炔酰菌胺、氟唑菌苯胺、吡噻菌胺、丙草胺、毒草胺、敌稗、氟胺磺隆、啶酰菌胺、呋虫胺、稻瘟酰胺、氟苯虫酰胺、氟噻草胺、氟酰胺、氟唑菌酰胺、吡虫啉、氯噻啉、噻虫嗪、烯肟菌胺、啶氧菌酯和肟菌酯中的至少一种。在本发明中,采用高效液相色谱-串联质谱检测所述待测液中低毒类农药含量的方法,优选包括以下步骤:
41.将所述待测液进行高效液相色谱-串联质谱检测,得到样品色谱图;
42.根据所述样品色谱图分别得到低毒类农药的峰面积;根据所述峰面积与低毒类农药的线性曲线,得到待测样品中低毒类农药的含量;所述低毒类农药的线性曲线为低毒类农药的色谱峰面积-质量浓度的线性曲线。
43.本发明将所述待测液进行高效液相色谱-串联质谱检测,得到样品色谱图。在本发明中,进行所述高效液相色谱-串联质谱检测采用的仪器优选为高效液相色谱(waters acquity h-class,美国waters公司)-三重四级杆质谱仪(ab sciex triple quad4500,美国ab sciex公司)。
44.在本发明中,所述高效液相色谱-串联质谱中高效液相色谱的检测条件包括:流动相体系为流动相a和流动相b,所述流动相a优选为甲醇,所述流动相b优选为乙酸铵水溶液,所述乙酸铵水溶液的浓度优选为2~10mmol/l,更优选为5mmol/l;所述流动相体系的流速优选为0.2~0.4ml/min,更优选为0.4ml/min;洗脱方式优选为梯度洗脱,所述梯度洗脱的程序优选为:
45.0.0~0.5min,流动相a的体积分数为2%;0.5~15.0min,流动相a的体积分数由2%增加至98%;15.0~17.0min,流动相a的体积分数维持在98%;17.0~17.1min,流动相a的体积分数由98%降低至2%;17.1~20.0min,流动相a的体积分数维持在2%。
46.在本发明中,所述高效液相色谱的检测条件还包括:色谱柱优选为acquity uplc hss t3色谱柱(100
×
2.1mm,1.8μm),柱温优选为40℃;进样量优选为3μl。
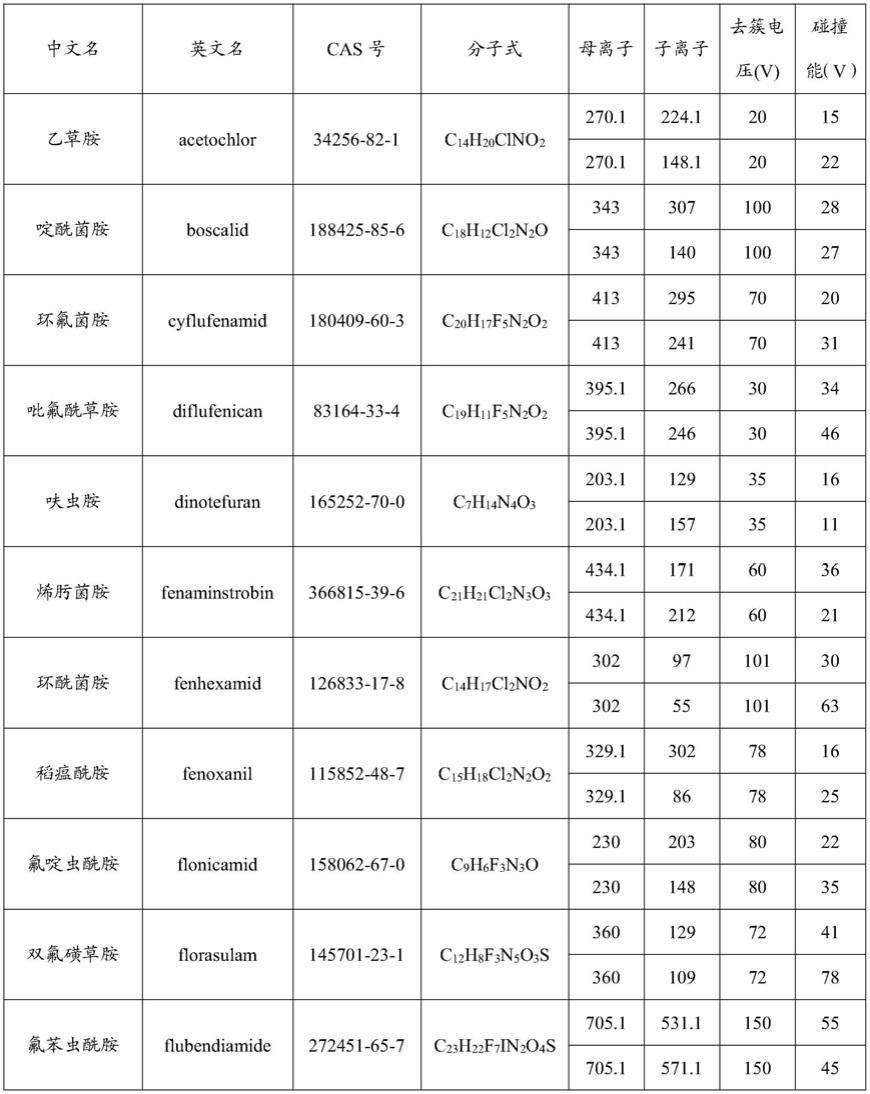
47.在本发明中,所述高效液相色谱-串联质谱中质谱的检测条件包括:离子源优选为电喷雾电离源(esi);检测方式优选为正离子模式;离子化电压(is)优选为4000~5500v,更优选为5500v;离子源温度优选为400~550℃,更优选为500~550℃;气帘气(cur)优选为空气,所述气帘气的压强优选为20~30psi,更优选为25~30psi;喷雾气(gs1)优选为空气,所述喷雾气的压强优选为40~50psi,更优选为45~50psi;辅助加热气(gs2)优选为空气,所述辅助加热气的压强优选为40~50psi,更优选为45~50psi;碰撞气(cad)优选为氮气,所述碰撞气的压强优选为4~7psi,更优选为6~7psi;所述31种低毒类农药的质谱多反应监测(mrm)参数优选如表1所示:
48.表1 31种低毒类农药的mrm参数
49.50.[0051][0052]
得到样品色谱图后,本发明根据所述样品色谱图得到低毒类农药的峰面积;根据所述峰面积与低毒类农药的线性曲线,得到待测样品中低毒类农药的含量;所述低毒类农药的线性曲线为低毒类农药的色谱峰面积-质量浓度的线性曲线。在本发明中,以建立31种低毒类农药的线性曲线为例,所述31种低毒类农药的线性曲线的建立方法,优选包括以下步骤:
[0053]
配制含有31种低毒类农药的混合标准中间液,所述混合标准中间液的浓度优选为5μg/ml;取适量体积的混合标准中间液,采用解吸附液对所述混合标准中间液进行稀释,分别得到浓度为1ng/ml、2ng/ml、5ng/ml、10ng/ml、50ng/ml和100ng/ml的混合标准液,然后按照上述条件对所述混合标准液进行高效液相色谱-串联质谱检测,得到31种低毒类农药的色谱峰,将所述色谱峰的峰面积与所述混合标准液中31种低毒类农药的质量浓度进行线性拟合,得到31种低毒类农药的线性曲线。
[0054]
在本发明中,所述31种低毒类农药的线性曲线、线性范围和相关系数,如表2所示。
[0055]
表2 31种低毒类农药的线性曲线、线性范围和相关系数
[0056]
[0057][0058]
本发明根据所述样品色谱图中低毒类农药的峰面积与低毒类农药的线性曲线,得到待测液中低毒类农药的含量,进而可以得到待测样品中低毒类农药的含量。
[0059]
下面将结合本发明中的实施例,对本发明中的技术方案进行清楚、完整地描述。显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
[0060]
实施例1
[0061]
将0.916g naoh、1.808g naalo2和33.975g四乙基氢氧化胺溶解在42.725g h2o中,在搅拌条件下加入23.0g硅胶粉,搅拌2h后转移到聚四氟乙烯衬里不锈钢高压釜中,在145℃条件下进行水热反应48h;自然冷却至室温后过滤,将得到的固体产物用去离子水洗涤后在100℃条件下干燥12h,然后在空气氛围中于550℃条件下煅烧6h,得到na-beta型分子筛;
[0062]
将na-beta型分子筛置于1mol/lnh4cl水溶液中,所述na-beta型分子筛和nh4cl水溶液的固液比为1g:50ml,在85℃条件下进行离子交换2h;自然冷却至室温后过滤,将得到的固体产物用去离子水洗涤后在100℃条件下干燥12h,然后在空气氛围中于550℃条件下煅烧6h,得到h-beta型分子筛,硅铝摩尔比为26,粒度为50~100μm。
[0063]
实施例2
[0064]
1.1仪器和试剂
[0065]
高效液相色谱(waters acquity h-class,美国waters公司)-三重四级杆质谱仪(ab sciex triple quad 4500,美国ab sciex公司),waters acquity uplc hss t3色谱柱(100
×
2.1mm,1.8μm,美国waters公司),mx-f涡动混合器(中国dragonlab公司),5415d离心机(美国eppendorf公司),milli-q净水系统(美国millipore公司),0.22μm尼龙过滤器(中国上海安谱公司)。
[0066]
浓度为100μg/ml的经认证的31种农药乙草胺、苯并烯氟菌唑、吡氟酰草胺、环酰菌胺、氟啶虫酰胺、双氟磺草胺、唑嘧磺草胺、氟吡菌胺、吡唑萘菌胺、双炔酰菌胺、氟唑菌苯胺、吡噻菌胺、丙草胺、毒草胺、敌稗、啶酰菌胺、呋虫胺、稻瘟酰胺、氟苯虫酰胺、氟噻草胺、氟酰胺、氟唑菌酰胺、吡虫啉、氯噻啉、烯肟菌胺、啶氧菌酯(以上标准品溶剂为甲醇)、氟吡菌酰胺、氟胺磺隆(以上标准品溶剂为乙腈)、环氟菌胺、噻虫嗪、肟菌酯(以上标准品溶剂为丙酮)的标准溶液,购自中国天津阿尔塔科技有限公司,色谱纯乙腈和甲醇购自美国merck
公司;色谱纯甲酸和乙酸铵购自上海安谱实验科技有限公司;实验室用水为一级水。
[0067]
1.2配制混合标准溶液
[0068]
将31种标准溶液用甲醇稀释成5μg/ml的混合标准中间液,于4℃避光保存,每两个月重新配制一次。
[0069]
1.3建立标准曲线
[0070]
取适量体积的混合标准中间液,采用甲酸-乙腈-甲醇混合液(甲酸的体积分数为0.5%,乙腈与甲醇的体积比为3:1)将所述混合标准中间液稀释至1ml,分别得到浓度为1ng/ml、2ng/ml、5ng/ml、10ng/ml、50ng/ml和100ng/ml的混合标准液,进行lc-ms/ms检测,其中,液相色谱检测条件具体如下:
[0071]
流动相体系为流动相a和流动相b,所述流动相a为甲醇,流动相b为浓度为5mmol/l的乙酸铵水溶液;所述流动相体系的流速为0.4ml/min;洗脱方式为梯度洗脱,梯度洗脱程序:0.0~0.5min,所述流动相a的体积分数为2%;0.5~15.0min,所述流动相a的体积分数由2%增加至98%;15.0~17.0min,所述流动相a的体积分数维持在98%;17.0~17.1min,所述流动相a的体积分数由98%降低至2%;17.1~20.0min,所述流动相a的体积分数维持在2%;色谱柱为acquity uplc hss t3柱(100
×
2.1mm,1.8μm);柱温为40℃;进样量为3μl;
[0072]
质谱检测条件具体如下:
[0073]
离子源为电喷雾电离源(esi),采用正离子模式;离子化电压(is)为5500v;离子源温度(tem)为550℃;气帘气(cur)为空气,压强为30psi;喷雾气(gs1)为空气,压强为50psi,辅助加热气(gs2)为空气,压强为50psi;碰撞气(cad)为氮气,压强为7psi;mrm参数如表1所示。
[0074]
lc-ms/ms检测过程中按混合标准溶液浓度从小到大依次进行lc-ms/ms检测,以峰面积(y)对质量浓度(x,ng/ml)进行线性拟合,得到线性曲线、线性范围和相关系数,结果如表2所示。
[0075]
实施例3
[0076]
水中农药的加标回收率试验,具体如下:
[0077]
在水中分别加入适量各农药的混合标准中间液,配制成浓度分别为0.08ng/ml、0.4ng/ml和2ng/ml的水溶液;分别将50ml水溶液置于塑料离心管中,加入50mg实施例1制备的h-beta型分子筛,密封后在室温(25℃)、2000rpm条件下涡旋吸附20min,然后在室温、5000rpm条件下离心5min,弃去全部上清液,向所得剩余物中加入2ml解吸附液,在室温条件下依次进行涡旋解吸附和超声解吸附,所述解吸附液为甲酸-乙腈-甲醇混合液,所述解吸附液中甲酸的体积分数为0.5%,乙腈与甲醇的体积比为3:1;所述涡旋解吸附的转速为2000rpm,时间为5min;所述超声解吸附的超声时间为5min;之后在5000rpm条件下离心3min,取上清液经0.22μm尼龙过滤器过滤后,按照实施例2中条件进行lc-ms/ms检测,每个水平6次平行测定,结果如表3和表4所示。其中,每个水平进行6次平行实验来获取方法的准确性(回收率)和日内精度rsd,日间精度rsd由每个水平的6次平行在不同天进行的三个添加回收实验测定的18组数据计算而得;检出限lod由高效液相色谱-三重四级杆质谱仪在添加浓度下的信噪比决定,被定义为信噪比大于3时的最低检出浓度,loq为信噪比大于10时的最低检出浓度。
[0078]
表3在水中农药的加标回收率(jb-0.08ng/ml和jb-0.4ng/ml)
[0079]
[0080][0081]
表4在水中农药的加标回收率(jb-2ng/ml)、检出限(lod)和定量限(loq)
[0082]
[0083][0084]
由表3和表4可知,在水中31种新型低毒农药做加标回收实验,浓度分别为0.08ng/ml、0.4ng/ml和2ng/ml时,31种农药在0.08ng/ml平均回收率为62.3~91.8%,相对标准偏差(rsd)为2.0~14.2%;在0.4ng/ml平均回收率为62.2~106.9%,相对标准偏差(rsd)为1.7~16.0%;在2ng/ml平均回收率为65.6~106.4%,相对标准偏差(rsd)为4.3~17.5%。结果显示,试验回收率和相对标准偏差均可以满足分析方法要求。
[0085]
实施例4
[0086]
采用图1所示流程对7个实际水样品(来源于城市河流和稻田水)中农药进行检测,具体如下:
[0087]
准确量取50ml实际水样品置于塑料离心管中,加入50mg实施例1制备的h-beta型分子筛,密封后在室温(25℃)、2000rpm条件下涡旋吸附20min,然后在室温、5000rpm条件下离心5min,弃去全部上清液,向所得剩余物中加入2ml解吸附液,在室温条件下依次进行涡旋解吸附和超声解吸附,所述解吸附液为甲酸-乙腈-甲醇混合液,所述解吸附液中甲酸的体积分数为0.5%,乙腈与甲醇的体积比为3:1;所述涡旋解吸附的转速为2000rpm,时间为5min;所述超声解吸附的时间为5min;之后在5000rpm条件下离心3min,取上清液经0.22μm尼龙过滤器过滤后,按照实施例2中条件进行lc-ms/ms检测。结果显示,吡虫啉在城市河流和稻田水中的7个样品均有检出,但含量低于定量限0.08ng/ml;稻瘟酰胺在4个来自稻田水的样品中被发现,其中两个稻田水样品(1号和2号)检出含量低于定量限0.08ng/ml,另外两个样品(3号和4号)检出值分别为0.11ng/ml和0.09ng/ml,3号样品和4号样品还有呋虫胺也被同时检出,但测定值低于定量限。
[0088]
由以上实施例可知,本发明基于h-beta型分子筛建立了分散固相萃取结合高效液
相色谱-串联质谱检测31种低毒类农药含量的方法,针对50ml水样品,h-beta型分子筛用量仅50mg,相比常见的固相萃取技术,所使用的吸附材料量较少,降低了成本;所使用的解吸附液量较少,仅为2ml;且测定的农药种类更多,范围更广,可满足水中31种低毒类农药的痕量检测分析。
[0089]
以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1