一种气相色谱-三重四极杆质谱法测定植物源性产品中草枯醚和氟除草醚残留量的方法与流程
1.本发明属于植物源性产品中草枯醚和氟除草醚残留量的测定技术领域,具体涉及一种气相色谱-三重四极杆质谱法测定植物源性产品中草枯醚和氟除草醚残留量的方法。
背景技术:
2.二苯醚类除草剂是一种原卟啉原氧化酶(protox)抑制剂,具有作用快,不影响作物产量,对后茬作物较安全等优点,广泛用于防除水稻、玉米、大豆、花生等作物的防除阔叶杂草。除草醚是最早合成的二苯醚类除草剂,之后在除草醚结构基础上添加各种取代基(羟基、硝基、卤基、烷氧基、酰基等)合成其他品种;氟除草醚就是在除草醚结构基础上添加卤基而得,其除草效果远优于除草醚而得到广泛使用。草枯醚是继除草醚之后合成的,曾大量使用而后因发现其具有致癌性而停止生产和使用,一些发达国家对草枯醚制定了严格的残留限量标准。草枯醚和氟除草醚的分子结构中均含有两个苯环,使得其水溶性小,脂溶性高,极易被土壤中的有机质吸附,从而在土壤中长期存留;此外,可经迁移、转化进入生物体内,在生物体内富集,对生物具有生物蓄积性、高毒性、潜在致癌性及内分泌干扰性。我国食品安全国家标准gb 2763-2021《食品安全国家标准食品中农药最大残留限量》对草枯醚和氟除草醚制定了蔬菜、水果、谷物、油料、茶叶、调味料和药用植物等植物性产品中的临时限量(均为0.01mg/kg),但目前并无成熟可用的植物性产品中草枯醚和氟除草醚定量检测方法,所以建立一种快速、灵敏、可靠的植物性产品草枯醚和氟除草醚的定量检测方法对于植物性产品中草枯醚和氟除草醚残留监控,以及风险评估等相关研究工作提供技术支持具有重要的意义。文献报道的草枯醚测定方法主要有使用气相色谱法、液相色谱法、气质联用法、液相色谱串联质谱法等,检测对象比较单一,主要为水、土壤和大豆等;氟除草醚的检测方法鲜有报道。
技术实现要素:
3.本发明的目的是针对现有的问题,提供了一种气相色谱-三重四极杆质谱法测定植物源性产品中草枯醚和氟除草醚残留量的方法。
4.本发明是通过以下技术方案实现的:
5.一种气相色谱-三重四极杆质谱法测定植物源性产品中草枯醚和氟除草醚残留量的方法,包括如下步骤:
6.s1、仪器、试剂与材料准备:
7.无水硫酸镁、氯化钠、乙腈、丙酮和正己烷均为分析纯,购于国药集团化学试剂有限公司;
8.乙酸乙酯,购于美国tedia公司;
9.乙二胺-n-丙基硅烷化硅胶(psa)、石墨化碳黑(gcb)和十八烷基键合硅胶(c
18
)购于上海安谱实验科技股份有限公司;
10.草枯醚购于坛墨质检科技股份有限公司,氟除草醚购于上海安谱璀世标准技术服务有限公司;
11.thermo 1300-tsq9000三重四极杆气质联用仪购自美国赛默飞世尔科技:配备电子轰击源;
12.s2、样品前处理:
13.s201、将蔬菜水果样品切碎匀浆,将谷物、茶叶、油料、调味料、药用植物等样品粉碎后充分混匀;
14.s202、称取10g蔬菜水果试样于50ml塑料离心管中;
15.s203、称取5g谷物、茶叶、油料、调味料、药用植物试样于50ml塑料离心管中,加入10ml饱和食盐水浸泡30min;
16.s204、加入4g氯化钠、10ml提取溶剂,一颗陶瓷均质子,漩涡振荡提取10min;4000r/min离心3min;
17.s205、准确吸取1.5ml上清液于2ml聚丙烯离心管,涡旋混合1min,14000r/min离心3min,取上清液过0.22μm有机滤膜,用于测定;
18.s3、标准溶液配制:
19.s301、准确称取草枯醚和氟除草醚标准品,分别用乙酸乙酯配制成质量浓度为1000mg/l的标准储备液;
20.s302、分别吸取的上述标准储备液,用乙酸乙酯配制成10mg/l的混合标准中间液;
21.s303、吸取混合标准中间液,用空白样品提取液稀释成质量浓度为2.5μg/l、10μg/l、50μg/l、200μg/l和500μg/l的基质混合标准工作溶液;
22.s4、基质效应的探究:
23.在吸附剂用量的优化中可知,草枯醚和氟除草醚在不同基质中表现出不同程度的基质效应,且通过分散固相萃取净化的方式无法将其回收率修正至合理范围,因此有必要对基质效应进行探究,以确保分析方法的准确性,按照s2前处理方法制备空白基质溶液,按照s3方法配制标准工作溶液和基质匹配标准工作溶液上机检测,对净化后基质效应进行评估;
24.s5、仪器条件:
25.柱箱升温程序:70℃保持0min,然后以25℃/min升温至230℃,保持0min;最后以10℃/min升温至310℃,保持2min;
26.载气:氦气,纯度≥99.999%,恒流模式,流量为1.0ml/min;
27.进样口温度:280℃;
28.进样量:1μl;
29.进样方式:不分流进样;
30.电子轰击源:70ev;离子源温度:300℃;
31.传输线温度:280℃;
32.溶剂延迟:4min;
33.采用多反应监测模式进行检测;
34.s6、实际样品检测:
35.对市场采购的上海青、芹菜、黄瓜、胡萝卜、姜、苹果、橙、大米、玉米、花生、茶叶、孜
然和三七样品各5个进行检测。
36.进一步地,步骤s202中所述的蔬菜水果和步骤s203中所述的谷物、茶叶、油料、调味料、药用植物称取时均精确至0.01g。
37.进一步地,步骤s204中还包括:
38.s2041、提取溶剂的选择:实验选择体积比为7:3的正己烷-丙酮、丙酮、乙酸乙酯、乙腈为提取溶剂,将添加有0.1mg/kg草枯醚和氟除草醚的芹菜、苹果、大米、花生、茶叶、孜然和三七样品按照s2进行提取离心后不净化直接上机检测,使用乙腈提取时提取液氮吹至干后用丙酮复溶后上机,使用溶剂标准工作曲线进行校正,每种样品平行测定3次。
39.进一步地,步骤s205中所述的聚丙烯离心管填装有净化试剂、吸附剂。
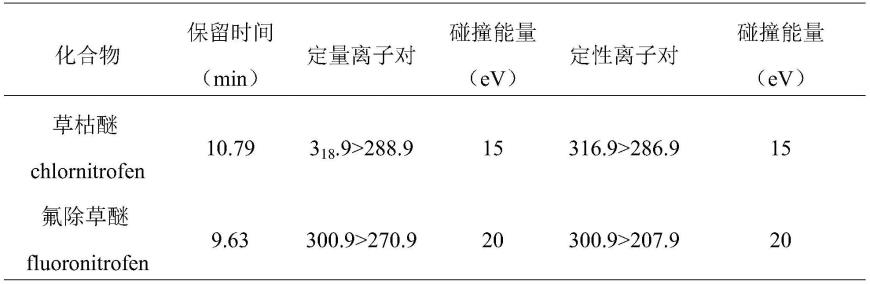
40.进一步地,还包括净化试剂的选择和吸附剂用量的优化。
41.进一步地,步骤s303中所述的空白样品提取液按照步骤s2的方法进行处理所得。
42.进一步地,步骤s5中还包括:
43.s501、质谱条件的选择:选择10.0mg/l的草枯醚和氟除草醚乙酸乙酯溶液,在质量数200~400范围内,对草枯醚和氟除草醚进行全扫描;选择质量数较大、响应较高的碎片离子进行二级碎裂,采用仪器自带软件auto srm优化碰撞电压;
44.s502、色谱柱的选择:实验对比了规格为30m
×
0.25mm
×
0.25μm的tg-5silms和规格为30m
×
0.25mm
×
0.25μm的tg-1701ms两种色谱柱对于草枯醚和氟除草醚的分离效果。
45.进一步地,步骤s5中所述的多反应监测模式:草枯醚和氟除草醚分别选择一个定量离子对、一个定性离子对,草枯醚的保留时间10.79min,定量离子对3
18
.9》288.9,碰撞能量15ev,定性离子对316.9》286.9,碰撞能量15ev;氟除草醚的保留时间9.63min,定量离子对300.9》270.9,碰撞能量20ev,定性离子对300.9》207.9,碰撞能量20ev。
46.进一步地,步骤s5中还包括:
47.s503、线性范围、相关系数和方法检测限的确定:用空白基质溶液配制浓度为2.5μg/l、10μg/l、50μg/l、200μg/l和500μg/l系列草枯醚和氟除草醚基质混合标准工作溶液,按仪器条件进行检测,以草枯醚和氟除草醚的质量浓度为横坐标,以草枯醚和氟除草醚峰面积y为纵坐标绘制基质标准工作曲线,得到线性方程和相关系数;在空白样品溶液中添加适量的标准溶液后上机测定,以s/n=10确定定量限;
48.s504、回收率和精密度的测定:分别对上海青、芹菜、黄瓜、胡萝卜、姜、苹果、橙、大米、玉米、花生、茶叶、孜然和三七空白样品进行了不同浓度的标准添加回收实验,每个加标水平测6次平行。
49.本发明相比现有技术具有以下优点:
50.1、本技术植物源性产品中残留的草枯醚和氟除草醚经乙酸乙酯提取,提取液经分散固相萃取净化,结合我国食品安全国家标准关于草枯醚和氟除草醚的残留限量要求,采用气相色谱-三重四极杆质谱仪建立了植物源性产品中草枯醚和氟除草醚的检测方法,建立的方法操作简单便捷,能满足植物源性产品中残留的草枯醚和氟除草醚检测要求。
51.2、本技术建立了气相色谱-三重四极杆质谱法同时测定植物源性产品中草枯醚和氟除草醚残留量的检测方法,方法具有较好的检测灵敏度和准确度。该方法操作简单、灵敏度高、回收率好,准确度高,能满足植物源性产品中残留的草枯醚和氟除草醚的检测需求和相关法规要求,可为植物源性产品中残留的草枯醚和氟除草醚风险监控提供有效的技术支
持。
附图说明
52.图1为草枯醚和氟除草醚提取离子色谱图;
53.图2为不同提取溶剂对草枯醚和氟除草醚的影响;
54.图3为不同吸附剂组合对加标空白样品提取回收率的影响;
55.图4为不同样品中草枯醚和氟除草醚的基质效应;
56.图5为上海青空白样品和加标样品提取离子色谱图;
57.图6为芹菜空白样品和加标样品提取离子色谱图;
58.图7为黄瓜空白样品和加标样品提取离子色谱图;
59.图8为胡萝卜空白样品和加标样品提取离子色谱图;
60.图9为姜空白样品和加标样品提取离子色谱图;
61.图10为苹果空白样品和加标样品提取离子色谱图;
62.图11为橙空白样品和加标样品提取离子色谱图;
63.图12为大米空白样品和加标样品提取离子色谱图;
64.图13为玉米空白样品和加标样品提取离子色谱图;
65.图14为花生空白样品和加标样品提取离子色谱图;
66.图15为茶叶空白样品和加标样品提取离子色谱图;
67.图16为孜然空白样品和加标样品提取离子色谱图;
68.图17为三七空白样品和加标样品提取离子色谱图。
具体实施方式
69.一种气相色谱-三重四极杆质谱法测定植物源性产品中草枯醚和氟除草醚残留量的方法,包括如下步骤:
70.s1、仪器、试剂与材料准备:
71.无水硫酸镁、氯化钠、乙腈、丙酮和正己烷均为分析纯,购于国药集团化学试剂有限公司;
72.乙酸乙酯,购于美国tedia公司;
73.乙二胺-n-丙基硅烷化硅胶(psa)、石墨化碳黑(gcb)和十八烷基键合硅胶(c
18
)购于上海安谱实验科技股份有限公司;
74.草枯醚购于坛墨质检科技股份有限公司,氟除草醚购于上海安谱璀世标准技术服务有限公司;
75.thermo 1300-tsq9000三重四极杆气质联用仪购自美国赛默飞世尔科技:配备电子轰击源;
76.s2、样品前处理:
77.s201、将蔬菜水果样品切碎匀浆,将谷物、茶叶、油料、调味料、药用植物等样品粉碎后充分混匀;
78.s202、称取10g蔬菜水果(精确至0.01g)试样于50ml塑料离心管中;
79.s203、称取5g谷物、茶叶、油料、调味料、药用植物(精确至0.01g)试样于50ml塑料
离心管中,加入10ml饱和食盐水浸泡30min;
80.s204、加入4g氯化钠、10ml提取溶剂,一颗陶瓷均质子,漩涡振荡提取10min;4000r/min离心3min;
81.s2041、提取溶剂的选择:实验选择体积比为7:3的正己烷-丙酮、丙酮、乙酸乙酯、乙腈为提取溶剂,将添加有0.1mg/kg草枯醚和氟除草醚的芹菜、苹果、大米、花生、茶叶、孜然和三七样品按照s2进行提取离心后不净化直接上机检测,使用乙腈提取时提取液氮吹至干后用丙酮复溶后上机,使用溶剂标准工作曲线进行校正,每种样品平行测定3次;得到提取溶液类型-平均回收率关系图如图2所示。
82.由图2可知:正己烷-丙酮(v:v,7:3)、丙酮、乙酸乙酯和乙腈为提取溶剂时,7种基质中草枯醚和氟除草醚的提取回收率均大于100%,均可以满足检测方法要求;7种基质在使用丙酮为提取溶剂时提取回收率最高,其次是正己烷-丙酮(7:3,v:v),再次是乙腈和乙酸乙酯,乙腈略高于乙酸乙酯。研究结果表明丙酮及正己烷-丙酮(7:3,v:v)中回收率较高,其原因是丙酮极性较强并能与水互溶,其对农药残留的提取效率较高。但丙酮作为提取溶剂时会将更多的植物样品中色素和油脂等共提取物提取出来,提取液颜色相对乙酸乙酯和乙腈更深,为下一步净化操作带来困难,所以前处理提取溶剂选择乙腈或者乙酸乙酯比较合适。因使用乙腈提取时需要进行氮吹操作,考虑便捷性,最终选择使用乙酸乙酯为提取溶剂。
83.s205、准确吸取1.5ml上清液于2ml聚丙烯离心管(填装有净化试剂、吸附剂),涡旋混合1min,14000r/min离心3min,取上清液过0.22μm有机滤膜,用于测定;
84.s2051、净化试剂的选择;
85.常用的quechers净化剂有gcb、c
18
、psa和nh2吸附剂等,其中psa和nh2吸附剂的吸附作用机理相似,二者均具有弱阴离子交换能力,通过氢键与化合物产生作用,可以有效去除样品中的有机酸、极性色素、脂肪酸、糖类以及其他能形成氢键的成分;c
18
可去除如挥发油、萜类、脂类等非极性化合物,gcb能去除色素、类胡萝卜素、类固醇和平面结构杂质的干扰,无水硫酸镁可以去除样液中的水分,实验考察了c
18
、gcb、psa和mgso4四种净化试剂对0.2mg/l草枯醚和氟除草醚混合标准溶液进行吸附后的回收率,实验结果表明,四种净化试剂对草枯醚和氟除草醚的平均吸附回收率都在90~110%之间,均能满足分析方法的要求;
86.s2052、吸附剂用量的优化:
87.植物源性样品种类繁多,基质也比较复杂,所以使用分散固相萃取净化时需要考虑两种或多种吸附净化剂,以较好的除去样品中的色素、脂类等干扰仪器分析的杂质。实验复配了含量不同的三组吸附剂组合(ⅰ:25mg psa+25mg gcb+25mg c
18
+50mg mgso4,ⅱ:50mg psa+50mg gcb+50mg c
18
+50mg mgso4,ⅲ:125mg psa+125mg gcb+125mg c
18
+50mg mgso4)对蔬菜、水果、谷物、油料、茶叶、调味料和药用植物的代表性基质样品芹菜、苹果、大米、花生、茶叶、孜然和三七空白样品0.20mg/kg加标样提取液1.5ml进行净化;提取净化实验步骤按照s2进行,考察不同组合吸附剂的净化效果和添加回收率。结果如图3所示,随着吸附剂用量的逐渐增加,草枯醚和氟除草醚的回收率均出现下降,但仅在净化试剂用量较大的组合(ⅲ:125mg psa+125mg gcb+125mg c
18
+50mg mgso4)下花生基质中草枯醚和氟除草醚的平均回收率能满足gb/t 27404-2008《实验室质量控制规范食品理化检测》对回收率(80%-110%)要求;其他基质中净化后基质效应仍较明显,回收率并无法满足分析方法要求的水
平;为了提高分析方法的准确性,仍然需要采取其他方式对不同基质中草枯醚和氟除草醚的基质效应进行补偿。综合考虑净化效果、成本、便捷性、仪器分析时基线干扰,选择50mg psa+50mg gcb+50mg c
18
+50mg mgso4组合对样品进行净化。
88.s3、标准溶液配制:
89.s301、准确称取草枯醚和氟除草醚标准品,分别用乙酸乙酯配制成质量浓度为1000mg/l的标准储备液;
90.s302、分别吸取的上述标准储备液,用乙酸乙酯配制成10mg/l的混合标准中间液;
91.s303、吸取混合标准中间液,用空白样品提取液稀释成质量浓度为2.5μg/l、10μg/l、50μg/l、200μg/l和500μg/l的基质混合标准工作溶液;
92.s4、基质效应的探究:
93.在吸附剂用量的优化中可知,草枯醚和氟除草醚在不同基质中表现出不同程度的基质效应,且通过分散固相萃取净化的方式无法将其回收率修正至合理范围,因此有必要对基质效应进行探究,以确保分析方法的准确性,按照s2前处理方法制备空白基质溶液,按照s3方法配制标准工作溶液和基质匹配标准工作溶液上机检测,对净化后基质效应进行评估;
94.s5、仪器条件:
95.s501、质谱条件的选择:选择10.0mg/l的草枯醚和氟除草醚乙酸乙酯溶液,在质量数200~400范围内,对草枯醚和氟除草醚进行全扫描;选择质量数较大、响应较高的碎片离子进行二级碎裂,采用仪器自带软件auto srm优化碰撞电压;草枯醚和氟除草醚定量离子对、定性离子对及碰撞能量见表1。0.1mg/l混合标准溶液中草枯醚和氟除草醚的提取离子色谱图见图1。
96.表1草枯醚和氟除草醚的保留时间、定量离子对、定性离子对和碰撞能量
[0097][0098]
s502、色谱柱的选择:实验对比了规格为30m
×
0.25mm
×
0.25μm的tg-5silms和规格为30m
×
0.25mm
×
0.25μm的tg-1701ms两种色谱柱对于草枯醚和氟除草醚的分离效果;
[0099]
结果显示草枯醚和氟除草醚在两种色谱柱上均能实现有效保留和分离,均有良好的峰型,但采用tg-5silms色谱柱时的峰面积明显高于tg-1701ms色谱柱,最终选择tg-5silms色谱柱作为本方法的色谱柱。
[0100]
s503、线性范围、相关系数和方法检测限的确定:用空白基质溶液配制浓度为2.5μg/l、10μg/l、50μg/l、200μg/l和500μg/l系列草枯醚和氟除草醚基质混合标准工作溶液,按仪器条件进行检测,以草枯醚和氟除草醚的质量浓度为横坐标,以草枯醚和氟除草醚峰面积y为纵坐标绘制基质标准工作曲线,得到线性方程和相关系数;在空白样品溶液中添加适
量的标准溶液后上机测定,以s/n=10确定定量限,相关数据见表2;
[0101]
表2草枯醚和氟除草醚的回归方程、相关系数、线性范围和定量限
[0102][0103]
s504、回收率和精密度的测定:分别对上海青、芹菜、黄瓜、胡萝卜、姜、苹果、橙、大米、玉米、花生、茶叶、孜然和三七空白样品进行了不同浓度的标准添加回收实验,每个加标水平测6次平行;
[0104]
表3草枯醚和氟除草醚在不同样品中三个水平下的平均加标回收率和相对标准偏差(n=6)
[0105][0106][0107]
确定的仪器条件为:
[0108]
柱箱升温程序:70℃保持0min,然后以25℃/min升温至230℃,保持0min;最后以10℃/min升温至310℃,保持2min;
[0109]
载气:氦气,纯度≥99.999%,恒流模式,流量为1.0ml/min;
[0110]
进样口温度:280℃;
[0111]
进样量:1μl;
[0112]
进样方式:不分流进样;
[0113]
电子轰击源:70ev;离子源温度:300℃;
[0114]
传输线温度:280℃;
[0115]
溶剂延迟:4min;
[0116]
采用多反应监测模式进行检测;
[0117]
s5、实际样品检测:
[0118]
对市场采购的上海青、芹菜、黄瓜、胡萝卜、姜、苹果、橙、大米、玉米、花生、茶叶、孜然和三七样品各5个进行检测。上海青、芹菜、黄瓜、胡萝卜、姜、苹果、橙、大米、玉米、花生、茶叶、孜然和三七空白样品和加标样品提取离子色谱图如图5~17所示。
[0119]
本文建立了气相色谱-三重四极杆质谱法同时测定植物源性产品中草枯醚和氟除草醚残留量的检测方法,方法具有较好的检测灵敏度和准确度。该方法操作简单、灵敏度高、回收率好,准确度高,能满足植物源性产品中残留的草枯醚和氟除草醚的检测需求和相关法规要求,可为植物源性产品中残留的草枯醚和氟除草醚风险监控提供有效的技术支持。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1