一种耳聋左慈丸混合粉中多种成分含量的测定方法与流程
1.本发明属于中药成分检测的技术领域,涉及一种耳聋左慈丸混合粉中多种成分含量的测定方法,具体涉及一种耳聋左慈丸混合粉中8种成分:5-羟甲基糠醛、莫诺苷、马钱苷、芍药苷、1,2,3,4,6-o-五没食子酰葡萄糖、苯甲酸、苯甲酰芍药苷和丹皮酚的hplc检测方法。
背景技术:
2.耳聋左慈丸始载于清
·
凌奂的《饲鹤亭集方》,收录于历年版《中国药典》中,是中医治疗肝肾阴虚所致耳鸣、耳聋的经典名方,其组成由六味地黄丸配方添加竹叶柴胡和磁石而成,广泛的临床应用及其药物研究进展均证实。本品滋肾平肝,用于肝肾阴虚,耳鸣耳聋,头晕目眩,对老年性、药物性及突发性耳聋、头晕目眩、视物恍惚及白内障证属肝肾阴亏、虚火上炎者等均有显著的疗效。耳聋左慈丸混合粉是耳聋左慈丸生产加工过程中的重要中间体,该中间体由熟地、山药、山茱萸、牡丹皮、茯苓、泽泻、竹叶柴胡和磁石八味中药材粉碎混合而成。
3.处方中的熟地黄滋阴补肾、填精益髓,是为君药;山茱萸补养肝肾、山药补益脾阴,两药相配伍,辅助君药,滋养肝脾肾,共为臣药;山茱萸补养肝阴、山药补益脾阴,两药相配伍,共为臣药;泽泻利湿卸浊,防止熟地黄滋腻恋邪,茯苓健脾渗湿,牡丹皮清泻相火,竹叶柴胡疏肝解郁,磁石重镇平肝,此五药共为佐药。
4.2020版《中国药典》对耳聋左慈丸混合粉中的含量检测项采用hplc法对其中马钱苷及莫诺苷进行检测。但耳聋左慈丸混合粉中成分众多,需要对其活性成分进行更细致的研究。
技术实现要素:
5.鉴于以上所述现有技术的缺点,本发明的目的在于提供一种耳聋左慈丸混合粉中多种成分含量的测定方法,用于解决现有技术中缺乏对耳聋左慈丸混合粉中多种有效成分进行测定从而对耳聋左慈丸混合粉的质量进行有效控制的问题。
6.为实现上述目的及其他相关目的,本发明第一方面提供一种耳聋左慈丸混合粉中多种成分含量的测定方法,包括:将耳聋左慈丸混合粉加入溶剂溶解后超声提取、摇匀、静置、过滤,取续滤液获得的供试品溶液采用高效液相色谱法(hplc)进行检测,确定供试品溶液中8种指标成分:5-羟甲基糠醛、莫诺苷、马钱苷、芍药苷、1,2,3,4,6-o-五没食子酰葡萄糖、苯甲酸、苯甲酰芍药苷、丹皮酚的含量。
7.较佳地,所述5-羟甲基糠醛(5-hmf)的cas号为67-47-0,所述莫诺苷(morroniside)的cas号为25406-64-8,所述马钱苷(loganin)的cas号为18524-94-2,所述芍药苷(paeoniflorin)的cas号为23180-57-6,所述1,2,3,4,6-o-五没食子酰葡萄糖(1,2,3,4,6-penta-o-galloy-β-d-glucose)的cas号为14937-32-7,所述苯甲酸(benzoic acid)的cas号为65-85-0,所述苯甲酰芍药苷(benzoylpaeoniflorin)的cas号为38642-49-8,所
述丹皮酚(paeonol)的cas号为552-41-0。
8.较佳地,所述耳聋左慈丸混合粉呈粉末状。
9.较佳地,所述耳聋左慈丸混合粉在制备供试品溶液前要进行
60
co-γ射线辐照。
10.更优选地,所述
60
co-γ射线辐照的剂量为0-10kgy,优选为0kgy、3kgy、6kgy、10kgy中的一种,更优选为6kgy。当所述
60
co-γ射线辐照的剂量为0kgy时,即未进行辐射。
11.更优选地,所述
60
co-γ射线辐照的最大吸收剂量≤6kgy。
12.所述
60
co-γ射线辐照灭菌技术是指利用
60
co产生的γ射线杀灭物料表面及内部微生物的过程。由于中药多以根及根茎部入药,生长于菌种较为丰富、微生物复杂的土壤环境中,药材经净制和切制后,仍会携带一定量的微生物。尤其是直接打粉入药的中成药品种,对药材的初始含菌量要求较为严格否则会造成成品含菌量超标。一定辐射量的辐照可以有效降低中药材中微生物的含量,同时降低微生物负载,达到对中药灭菌的效果。因此,有必要对中药制剂原料药材的生药粉进行辐照灭菌,从而进一步降低微生物负载。
13.较佳地,所述耳聋左慈丸混合粉加入溶剂前要精密称重,所述耳聋左慈丸混合粉加入溶剂后也要精密称重。
14.较佳地,所述溶剂为70-90%甲醇。优选地,所述溶剂为80%甲醇。
15.上述70-90%甲醇为体积百分比为70-90%(v/v)的甲醇水溶液。上述80%甲醇为体积百分比为80%(v/v)的甲醇水溶液。
16.较佳地,所述耳聋左慈丸混合粉加入的质量g与溶剂加入的体积ml之比为1:15-25,优选为1:20。
17.较佳地,所述超声提取的时间为30-60分钟,优选为40-50分钟,更优选为45分钟。
18.较佳地,所述超声提取的功率为200-300w,优选为250w;所述超声提取的频率为30-50khz,优选为40khz。
19.较佳地,所述超声提取后要进去冷却,精密称重后用溶剂补足失重。所述冷却至室温,所述室温为20-30℃。
20.较佳地,所述静置片刻以使超声提取后的溶液分层。
21.较佳地,所述过滤为取上清液过滤膜,放弃初滤液后,取续滤液。
22.优选地,所述滤膜为0.45μm滤膜。
23.较佳地,所述采用高效液相色谱法(hplc)进行检测,包括以下步骤:
24.1)配制对照品溶液:将5-羟甲基糠醛、莫诺苷、马钱苷、芍药苷、1,2,3,4,6-o-五没食子酰葡萄糖、苯甲酸、苯甲酰芍药苷、丹皮酚的对照品,加入溶剂溶解并定容,配成对照品溶液;
25.2)样品检测:采用高效液相色谱法(hplc)分别检测供试品溶液和步骤1)中的对照品溶液,比较保留时间进行定性,采用外标法进行定量,确定供试品溶液中5-羟甲基糠醛、莫诺苷、马钱苷、芍药苷、1,2,3,4,6-o-五没食子酰葡萄糖、苯甲酸、苯甲酰芍药苷、丹皮酚的含量。
26.优选地,步骤1)中,所述对照品溶液采用逐级稀释制得。
27.更优选地,所述逐级稀释采用的对照品储备溶液中,5-羟甲基糠醛的含量为201.60μg/ml,莫诺苷的含量为90.00μg/ml,马钱苷的含量为102.80μg/ml,芍药苷的含量为126.04μg/ml,1,2,3,4,6-o-五没食子酰葡萄糖的含量为121.20μg/ml,苯甲酸的含量为
18.40μg/ml,苯甲酰芍药苷的含量为37.84μg/ml,丹皮酚的含量为164.04μg/ml。
28.优选地,步骤1)中,所述对照品溶液中5-羟甲基糠醛的含量范围为0.202-4.032μg/ml,莫诺苷的含量范围为0.090-1.800μg/ml,马钱苷的含量范围为0.103-2.056μg/ml,芍药苷的含量范围为0.126-2.521μg/ml,1,2,3,4,6-o-五没食子酰葡萄糖的含量范围为0.121-2.424μg/ml,苯甲酸的含量范围为0.018-0.368μg/ml,苯甲酰芍药苷的含量范围为0.038-0.756μg/ml,丹皮酚的含量范围为0.164-3.281μg/ml。
29.优选地,步骤1)中,所述溶剂为70-90%甲醇。更优选地,所述溶剂为80%甲醇。
30.上述70-90%甲醇为体积百分比为70-90%(v/v)的甲醇水溶液。上述80%甲醇为体积百分比为80%(v/v)的甲醇水溶液。
31.优选地,步骤2)中,所述高效液相色谱法(hplc)中,检测器为二极管阵列检测器(dad)。
32.优选地,步骤2)中,所述高效液相色谱法中的色谱柱为ods色谱柱。
33.更优选地,所述高效液相色谱法中的色谱柱为dikma platisil ods色谱柱(柱长为250mm,内径为4.6mm,填料粒径为5μm)。
34.优选地,步骤2)中,所述高效液相色谱法中的检测波长为230-250nm。更优选地,所述检测波长为240nm。
35.优选地,步骤2)中,所述高效液相色谱法中柱温为25-30℃。更优选地,所述高效液相色谱法中柱温为28℃。
36.优选地,步骤2)中,所述高效液相色谱法中的流速为0.5-1.5ml/min。更优选地,所述高效液相色谱法中的流速为1.0ml/min。
37.优选地,步骤2)中,所述高效液相色谱法中的进样量为5-15μl。更优选地,所述高效液相色谱法中的进样量为10μl。
38.优选地,步骤2)中,所述高效液相色谱法中,流动相为乙腈-0.05-0.15%磷酸水溶液,其中,a相为乙腈,b相为0.05-0.15%磷酸水溶液;分析时间为90min;梯度洗脱。更优选地,所述高效液相色谱法中,流动相为乙腈-0.10%磷酸水溶液,其中,a相为乙腈,b相为0.10%磷酸水溶液;分析时间为90min;梯度洗脱。
39.上述0.05-0.15%磷酸水溶液为体积百分数为0.05-0.15%的磷酸水溶液。上述0.10%磷酸水溶液为体积百分数为0.10的磷酸水溶液。
40.更优选地,所述梯度洗脱的具体程序为:
41.0~12min,a相:b相体积比为5:95-9:91;
42.12~31min,a相:b相体积比为9:91-19:81;
43.31~35min,a相:b相体积比为19:81-19:81;
44.35~50min,a相:b相体积比为19:81-27:73;
45.50~70min,a相:b相体积比为27:73-69:31;
46.70~80min,a相:b相体积比为69:31-81:19;
47.80~90min,a相:b相体积比为81:19-95:5。
48.优选地,步骤2)中,所述外标法包括以下步骤:
49.a)按步骤1)制备一系列不同浓度的对照品溶液,分别进行hplc检测,获得5-羟甲基糠醛、莫诺苷、马钱苷、芍药苷、1,2,3,4,6-o-五没食子酰葡萄糖、苯甲酸、苯甲酰芍药苷、
丹皮酚的色谱峰面积与对应5-羟甲基糠醛、莫诺苷、马钱苷、芍药苷、1,2,3,4,6-o-五没食子酰葡萄糖、苯甲酸、苯甲酰芍药苷、丹皮酚的含量之间线性关系,绘制相应的标准工作曲线,分别计算得到5-羟甲基糠醛、莫诺苷、马钱苷、芍药苷、1,2,3,4,6-o-五没食子酰葡萄糖、苯甲酸、苯甲酰芍药苷、丹皮酚的标准工作曲线的回归方程;
50.b)将供试品溶液进行hplc检测,将获得的5-羟甲基糠醛、莫诺苷、马钱苷、芍药苷、1,2,3,4,6-o-五没食子酰葡萄糖、苯甲酸、苯甲酰芍药苷、丹皮酚的色谱峰面积,代入步骤a)中相应的5-羟甲基糠醛、莫诺苷、马钱苷、芍药苷、1,2,3,4,6-o-五没食子酰葡萄糖、苯甲酸、苯甲酰芍药苷、丹皮酚的标准工作曲线的回归方程,计算得到供试品溶液中5-羟甲基糠醛、莫诺苷、马钱苷、芍药苷、1,2,3,4,6-o-五没食子酰葡萄糖、苯甲酸、苯甲酰芍药苷、丹皮酚的含量。
51.更优选地,所述标准工作曲线中,以5-羟甲基糠醛、莫诺苷、马钱苷、芍药苷、1,2,3,4,6-o-五没食子酰葡萄糖、苯甲酸、苯甲酰芍药苷、丹皮酚的进样量(取样的对照品储备溶液的浓度确定,通过取不同进样量获得一系列不同浓度的对照品溶液)为横坐标(x轴),其相应5-羟甲基糠醛、莫诺苷、马钱苷、芍药苷、1,2,3,4,6-o-五没食子酰葡萄糖、苯甲酸、苯甲酰芍药苷、丹皮酚的色谱峰面积为纵坐标(y轴)。
52.本发明第二方面提供一种耳聋左慈丸混合粉中多种成分含量的测定方法在耳聋左慈丸混合粉的质量检测中的用途。
53.如上所述,本发明提供的一种耳聋左慈丸混合粉中多种成分含量的测定方法,采用优化条件的前处理和仪器检测方法,可以在同一次分析中对耳聋左慈丸混合粉中8种活性成分:5-羟甲基糠醛、莫诺苷、马钱苷、芍药苷、1,2,3,4,6-o-五没食子酰葡萄糖、苯甲酸、苯甲酰芍药苷、丹皮酚进行准确的定量定性检测。该种方法所测定的8种成分的标准曲线在各自范围内线性关系良好,平均加样回收率为97.38-103.05%,rsd均小于3.02%,重复性良好、精密度良好、准确度高、稳定性好,具有提高分析效率、降低人力物力等成本的优势,可真实反映耳聋左慈丸混合粉中多种指标成分的质量差异,完善耳聋左慈丸混合粉的质量控制体系。
附图说明
54.图1显示为本发明的耳聋左慈丸混合粉中8种成分的高效液相色谱图1a、1b,其中,图1a为8种成分的标准品的高效液相色谱图,图1b为8种成分的供试品的高效液相色谱图;图1a和1b中,1为5-羟甲基糠醛,2为莫诺苷,3为马钱苷,4为芍药苷,5为1,2,3,4,6-o-五没食子酰葡萄糖,6为苯甲酸,7为苯甲酰芍药苷,8为丹皮酚。
具体实施方式
55.下面结合具体实施例进一步阐述本发明,应理解,这些实施例仅用于说明本发明而不用于限制本发明的保护范围。
56.以下通过特定的具体实例说明本发明的实施方式,本领域技术人员可由本说明书所揭露的内容轻易地了解本发明的其他优点与功效。本发明还可以通过另外不同的具体实施方式加以实施或应用,本说明书中的各项细节也可以基于不同观点与应用,在没有背离本发明的精神下进行各种修饰或改变。
57.以下实施例使用的试剂和仪器如下:
58.1、试剂
59.耳聋左慈丸混合粉(批号分别为190401、190402、190403、190404、190405、190406、190407、190408、190409和1904010,上海和黄药业有限公司制备)。
60.5-羟甲基糠醛(批号19042521,纯度100%)、丹皮酚(批号18071943,纯度99.9%)、芍药苷(批号18051621,纯度》98%)、苯甲酰芍药苷(批号19031632,纯度99.3%)、1,2,3,4,6-o-五没食子酰葡萄糖(批号19032823,纯度98.2%)均购自上海同田生物技术股份有限公司;苯甲酸(批号5896,纯度99.7%)、莫诺苷(批号5312,纯度96.4%)、马钱苷(批号5651,纯度98.7%)均购自上海诗丹德生物技术有限公司。
61.甲醇(分析纯ar,国药集团化学试剂有限公司),乙醇(分析纯ar,上海波尔化学试剂有限公司),乙腈(色谱纯,美国tedia公司),磷酸(色谱纯,美国tedia公司),超纯水由milli-q超纯水处理系统制备。
62.2、仪器
63.agilent1260高效液相色谱仪(openlab cds 2.1色谱工作站,g1312b二元泵,g1322a自动脱气装置,g7116a柱温箱,g7115a dad检测器,g7129a自动进样器;美国安捷伦公司);sb-5200dtd型超声波清洗机(宁波新芝生物科技股份有限公司);电热鼓风干燥箱(上海一恒科学仪器有限公司);al204和xs205型分析电子天平(梅特勒-托利多仪器上海有限公司);milli-q advantage a10超纯水系统(德国默克密理博公司)。
64.实施例1
65.1、供试品溶液的制备
66.取耳聋左慈丸混合粉样品。在剂量为6kgy下进行
60
co-γ射线辐照,
60
co-γ射线辐照的最大吸收剂量≤6kgy。取上述耳聋左慈丸混合粉样品1.0g,精密称定,置于50ml的具塞锥形瓶中,精密加入80%甲醇20ml,密塞,称定重量,超声提取(功率250w,频率40khz)45min,放冷,再称定重量,用80%甲醇补足失重,摇匀,静置片刻,取上清液,过0.45μm滤膜,取续滤液,即得供试品溶液1#。
67.2、对照品溶液的制备
68.取5-羟甲基糠醛、莫诺苷、马钱苷、芍药苷、1,2,3,4,6-o-五没食子酰葡萄糖、苯甲酸、苯甲酰芍药苷、丹皮酚的对照品,置于同一100ml容量瓶中,用80%甲醇溶解并稀释定容至刻度,摇匀,配成对照品储备溶液。对照品储备溶液中,5-羟甲基糠醛的含量为201.60μg/ml,莫诺苷的含量为90.00μg/ml,马钱苷的含量为102.80μg/ml,芍药苷的含量为126.04μg/ml,1,2,3,4,6-o-五没食子酰葡萄糖的含量为121.20μg/ml,苯甲酸的含量为18.40μg/ml,苯甲酰芍药苷的含量为37.84μg/ml,丹皮酚的含量为164.04μg/ml。
69.再将对照品储备溶液精密称定,采用80%甲醇逐级稀释并定容,配成一系列不同浓度的对照品溶液。一系列不同浓度的对照品溶液中,5-羟甲基糠醛的含量范围为0.202-4.032μg/ml,莫诺苷的含量范围为0.090-1.800μg/ml,马钱苷的含量范围为0.103-2.056μg/ml,芍药苷的含量范围为0.126-2.521μg/ml,1,2,3,4,6-o-五没食子酰葡萄糖的含量范围为0.121-2.424μg/ml,苯甲酸的含量范围为0.018-0.368μg/ml,苯甲酰芍药苷的含量范围为0.038-0.756μg/ml,丹皮酚的含量范围为0.164-3.281μg/ml。
70.3、测定
71.采用高效液相色谱法(hplc)分别检测供试品溶液1#和一系列不同浓度的对照品溶液,比较保留时间进行定性,采用外标法进行定量。即将一系列不同浓度的对照品溶液分别进行hplc检测,获得5-羟甲基糠醛、莫诺苷、马钱苷、芍药苷、1,2,3,4,6-o-五没食子酰葡萄糖、苯甲酸、苯甲酰芍药苷、丹皮酚的色谱峰面积与对应5-羟甲基糠醛、莫诺苷、马钱苷、芍药苷、1,2,3,4,6-o-五没食子酰葡萄糖、苯甲酸、苯甲酰芍药苷、丹皮酚的浓度之间线性关系,绘制相应的标准工作曲线,分别计算得到5-羟甲基糠醛、莫诺苷、马钱苷、芍药苷、1,2,3,4,6-o-五没食子酰葡萄糖、苯甲酸、苯甲酰芍药苷、丹皮酚的标准工作曲线的回归方程。再将供试品溶液1#进行hplc检测,将获得的5-羟甲基糠醛、莫诺苷、马钱苷、芍药苷、1,2,3,4,6-o-五没食子酰葡萄糖、苯甲酸、苯甲酰芍药苷、丹皮酚的色谱峰面积,代入相应的5-羟甲基糠醛、莫诺苷、马钱苷、芍药苷、1,2,3,4,6-o-五没食子酰葡萄糖、苯甲酸、苯甲酰芍药苷、丹皮酚的标准工作曲线的回归方程,计算得到供试品溶液1#中5-羟甲基糠醛、莫诺苷、马钱苷、芍药苷、1,2,3,4,6-o-五没食子酰葡萄糖、苯甲酸、苯甲酰芍药苷、丹皮酚的浓度。
72.其中,高效液相色谱法包括以下检测条件:
73.检测器为二极管阵列检测器(dad);色谱柱为dikma platisil ods色谱柱(柱长为250mm,内径为4.6mm,填料粒径为5μm);检测波长为240nm;柱温为28℃;流速为1.0ml/min;进样量为10μl;流动相为乙腈-0.05-0.15%磷酸水溶液,其中,a相为乙腈,b相为0.05-0.15%磷酸水溶液;分析时间为90min;梯度洗脱。
74.梯度洗脱的具体程序为:
75.0~12min,a相:b相体积比为5:95-9:91;
76.12~31min,a相:b相体积比为9:91-19:81;
77.31~35min,a相:b相体积比为19:81-19:81;
78.35~50min,a相:b相体积比为19:81-27:73;
79.50~70min,a相:b相体积比为27:73-69:31;
80.70~80min,a相:b相体积比为69:31-81:19;
81.80~90min,a相:b相体积比为81:19-95:5。
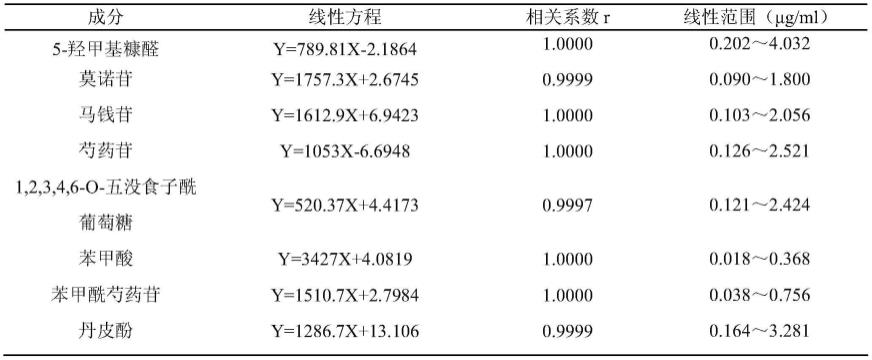
82.实施例2
83.1、供试品溶液的制备
84.取耳聋左慈丸混合粉样品。在剂量为3kgy下进行
60
co-γ射线辐照,
60
co-γ射线辐照的最大吸收剂量≤6kgy。取上述耳聋左慈丸混合粉样品1.0g,精密称定,置于50ml的具塞锥形瓶中,精密加入75%甲醇15ml,密塞,称定重量,超声提取(功率240w,频率30khz)50min,放冷,再称定重量,用75%甲醇补足失重,摇匀,静置片刻,取上清液,过0.45μm滤膜,取续滤液,即得供试品溶液2#。
85.2、对照品溶液的制备
86.取5-羟甲基糠醛、莫诺苷、马钱苷、芍药苷、1,2,3,4,6-o-五没食子酰葡萄糖、苯甲酸、苯甲酰芍药苷、丹皮酚的对照品,置于同一100ml容量瓶中,用75%甲醇溶解并稀释定容至刻度,摇匀,配成对照品储备溶液。再将对照品储备溶液精密称定,采用75%甲醇逐级稀释并定容,配成一系列不同浓度的对照品溶液。
87.对照品储备溶液和一系列不同浓度的对照品溶液中,5-羟甲基糠醛、莫诺苷、马钱
苷、芍药苷、1,2,3,4,6-o-五没食子酰葡萄糖、苯甲酸、苯甲酰芍药苷、丹皮酚的浓度范围同实施例1中步骤2。
88.3、测定
89.采用高效液相色谱法(hplc)分别检测供试品溶液2#和一系列不同浓度的对照品溶液,比较保留时间进行定性,采用外标法进行定量,得到供试品溶液2#中5-羟甲基糠醛、莫诺苷、马钱苷、芍药苷、1,2,3,4,6-o-五没食子酰葡萄糖、苯甲酸、苯甲酰芍药苷、丹皮酚的浓度。具体定量过程同实施例1中步骤3。
90.其中,高效液相色谱法包括以下检测条件:
91.检测器为二极管阵列检测器(dad);色谱柱为dikma platisil ods色谱柱(柱长为250mm,内径为4.6mm,填料粒径为5μm);检测波长为245nm;柱温为30℃;流速为0.5ml/min;进样量为5μl;流动相为乙腈-0.15%磷酸水溶液,其中,a相为乙腈,b相为0.15%磷酸水溶液;分析时间为90min;梯度洗脱。梯度洗脱的具体程序同实施例1中步骤3。
92.实施例3
93.1、供试品溶液的制备
94.取耳聋左慈丸混合粉样品。在剂量为10kgy下进行60co-γ射线辐照,
60
co-γ射线辐照的最大吸收剂量≤6kgy。取上述耳聋左慈丸混合粉样品1.0g,精密称定,置于50ml的具塞锥形瓶中,精密加入85%甲醇25ml,密塞,称定重量,超声提取(功率260w,频率50khz)40min,放冷,再称定重量,用85%甲醇补足失重,摇匀,静置片刻,取上清液,过0.45μm滤膜,取续滤液,即得供试品溶液3#。
95.2、对照品溶液的制备
96.取5-羟甲基糠醛、莫诺苷、马钱苷、芍药苷、1,2,3,4,6-o-五没食子酰葡萄糖、苯甲酸、苯甲酰芍药苷、丹皮酚的对照品,置于同一100ml容量瓶中,用85%甲醇溶解并稀释定容至刻度,摇匀,配成对照品储备溶液。再将对照品储备溶液精密称定,采用85%甲醇逐级稀释并定容,配成一系列不同浓度的对照品溶液。
97.对照品储备溶液和一系列不同浓度的对照品溶液中,5-羟甲基糠醛、莫诺苷、马钱苷、芍药苷、1,2,3,4,6-o-五没食子酰葡萄糖、苯甲酸、苯甲酰芍药苷、丹皮酚的浓度范围同实施例1中步骤2。
98.3、测定
99.采用高效液相色谱法(hplc)分别检测供试品溶液3#和一系列不同浓度的对照品溶液,比较保留时间进行定性,采用外标法进行定量,得到供试品溶液3#中5-羟甲基糠醛、莫诺苷、马钱苷、芍药苷、1,2,3,4,6-o-五没食子酰葡萄糖、苯甲酸、苯甲酰芍药苷、丹皮酚的浓度。具体定量过程同实施例1中步骤3。
100.其中,高效液相色谱法包括以下检测条件:
101.检测器为二极管阵列检测器(dad);色谱柱为dikma platisil ods色谱柱(柱长为250mm,内径为4.6mm,填料粒径为5μm);检测波长为235nm;柱温为25℃;流速为1.5ml/min;进样量为15μl;流动相为乙腈-0.05%磷酸水溶液,其中,a相为乙腈,b相为0.05%磷酸水溶液;分析时间为90min;梯度洗脱。梯度洗脱的具体程序同实施例1中步骤3。
102.实施例4
103.取实施例1中步骤1配制的供试品溶液1#和实施例1中步骤2配制的任一对照品溶
液,按实施例1中步骤3进行测定,比较保留时间进行定性,具体测试结果见图1。由图1可知,供试品溶液1#与对照品溶液的保留时间一致,各成分色谱峰峰型均较对称,分离度良好,理论塔板数均大于5000,系统适用性和专属性良好。
104.实施例5
105.对本发明的耳聋左慈丸混合粉样品中5-羟甲基糠醛、莫诺苷、马钱苷、芍药苷、1,2,3,4,6-o-五没食子酰葡萄糖、苯甲酸、苯甲酰芍药苷、丹皮酚的检测方法进行方法学验证,其性能指标结果如下。
106.1、检测方法的线性关系
107.精密称取实施例1中步骤2配制的对照品储备溶液1、2、5、10、15、20μl,按实施例1中步骤3的色谱条件进行测定,以各指标成分的峰面积为纵坐标(y轴),以对照品的进样量为横坐标(x轴),绘制标准曲线,计算获得5-羟甲基糠醛、莫诺苷、马钱苷、芍药苷、1,2,3,4,6-o-五没食子酰葡萄糖、苯甲酸、苯甲酰芍药苷、丹皮酚的标准回归方程、相关系数和线性范围,具体结果见表1。根据表1可知,8个指标成分在各自进样质量浓度范围内线性关系良好,说明本发明方法线性范围广,准确度高。
108.表1
[0109][0110]
2、稳定性
[0111]
取批号190401(0kgy-未辐照)的耳聋左慈丸混合粉样品,按实施例1中步骤1制备获得供试品溶液,按实施例1中步骤3的色谱条件,分别在0h、2h、4h、6h、8h、12h、24h进样分析,记录峰面积。结果表明,5-羟甲基糠醛、莫诺苷、马钱苷、芍药苷、1,2,3,4,6-o-五没食子酰葡萄糖、苯甲酸、苯甲酰芍药苷、丹皮酚的色谱峰面积在24h内的rsd均小于3%,表明供试品溶液在24h内检测对结果无影响,稳定性良好。
[0112]
3、精密度
[0113]
取实施例1中步骤2配制的任一对照品溶液,按实施例1中步骤3的色谱条件,分别连续进样分析6次,记录峰面积。结果表明,8个成分连续6次进样的峰面积rsd均小于2%,表明仪器精密度良好。
[0114]
4、重复性
[0115]
取批号为190401(0kgy-未辐照)的耳聋左慈丸混合粉样品,按实施例1中步骤1平行制备获得6份供试品溶液,按实施例1中步骤3的色谱条件进行测定,记录色谱图。结果表明,5-羟甲基糠醛、莫诺苷、马钱苷、芍药苷、1,2,3,4,6-o-五没食子酰葡萄糖、苯甲酸、苯甲
酰芍药苷、丹皮酚的色谱峰面积的rsd均小于3%,表明方法重复性良好。
[0116]
5、加标回收率
[0117]
精密称取9份已知浓度的耳聋左慈丸混合粉样品(批号190401 0kgy-未辐照)0.5g,精密称定,按三个不同浓度水平(1:0.5、1:1、1:1.5)加入实施例1中步骤2配制的任一对照品溶液。按实施例1中步骤1制备获得供试品溶液,并按实施例1中步骤3的色谱条件,分别进样分析,记录色谱图,计算各成分回收率,结果见表2。由表2可知,8个成分的平均加样回收率97.38-103.05%,rsd均小于3.02%,表明该测定方法准确度较好。
[0118]
表2
[0119]
检测成分平均加样回收率rsd5-羟甲基糠醛97.38%2.05%莫诺苷98.49%1.74%马钱苷100.95%1.00%芍药苷99.80%1.83%1,2,3,4,6-o-五没食子酰葡萄糖102.26%2.83%苯甲酸103.05%3.02%苯甲酰芍药苷98.99%1.40%丹皮酚99.32%1.12%
[0120]
实施例6
[0121]
取多批号的耳聋左慈丸混合粉,分别以剂量0、3、6、10kgy辐照处理后,按实施例1中步骤1制备获得供试品溶液,并按实施例1中步骤3的色谱条件,分别进样分析,分别计算辐照前后耳聋左慈丸混合粉中5-羟甲基糠醛、莫诺苷、马钱苷、芍药苷、1,2,3,4,6-o-五没食子酰葡萄糖、苯甲酸、苯甲酰芍药苷和丹皮酚的含量,结果见表3。
[0122]
表3各样品含量测定结果(mg
·
g-1
,n=10)
[0123][0124]
由表3可知,采用spss 20.0软件对
60
co辐照前后耳聋左慈丸混合粉中8个成分含量进行成组t检验分析,以考察不同辐照剂量组之间的各成分(5-羟甲基糠醛、莫诺苷、马钱苷、芍药苷、1,2,3,4,6-o-五没食子酰葡萄糖、苯甲酸、苯甲酰芍药苷和丹皮酚)含量差异是否具有统计学意义。结果其8种成分的p值均大于0.05,表明组间均数的差异不具有统计学意义,即这8种成分经不同剂量(3kgy、6kgy、10kgy)
60
co辐照后含量变化没有显著性差异,即本研究所用剂量的
60
co辐照灭菌不影响耳聋左慈丸混合粉中8种成分的含量。
[0125]
综上所述,本发明提供的一种耳聋左慈丸混合粉中多种成分含量的测定方法,重复性良好、精密度良好、准确度高、稳定性好,可真实反映耳聋左慈丸混合粉中多种指标成
分的质量差异,完善耳聋左慈丸混合粉的质量控制体系。所以,本发明有效克服了现有技术中的种种缺点而具高度产业利用价值。
[0126]
上述实施例仅例示性说明本发明的原理及其功效,而非用于限制本发明。任何熟悉此技术的人士皆可在不违背本发明的精神及范畴下,对上述实施例进行修饰或改变。因此,举凡所属技术领域中具有通常知识者在未脱离本发明所揭示的精神与技术思想下所完成的一切等效修饰或改变,仍应由本发明的权利要求所涵盖。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1