一种十全大补丸混合粉指纹图谱及多成分的检测方法与流程
1.本发明属于中药成分检测的技术领域,涉及一种十全大补丸混合粉指纹图谱及多成分的检测方法,具体涉及一种十全大补丸混合粉指纹图谱及其8种主要有效成分:5-羟甲基糠醛、芍药苷、甘草苷、阿魏酸、1,2,3,4,6-o-五没食子酰葡萄糖、桂皮醛、甘草酸、藁本内酯的检测方法。
背景技术:
2.十全大补丸是传统的益气补血类中药制剂,收载于2020版《中国药典》,处方由黄芪、党参、熟地黄、白术、茯苓、当归、白芍、川芎、肉桂、炙甘草,共十味中药组成,有温补气血之功效,临床多用于气血两虚、面色苍白、气短心悸、四肢不温等症。
3.十全大补丸混合粉是十全大补丸生产加工过程中的主要中间体之一,该中间体由炙黄芪、党参、熟地黄、炒白术、茯苓、当归、酒白芍、川芎、肉桂和炙甘草十味中药材混合粉碎而成。方中药味组成多,化学成分复杂,目前缺乏准确有效的检验方法。为了有效监控十全大补丸混合粉质量,有必要建立一套灵敏高效,简便快速的分析方法,既能展现全方复杂的物质基础,又能对重点药效成分进行定量研究,作为十全大补丸中间体质量控制的主要手段。
技术实现要素:
4.鉴于以上所述现有技术的缺点,本发明的目的在于提供一种十全大补丸混合粉指纹图谱及多成分的检测方法,采用优化条件的前处理及高效液相色谱方法,建立了十全大补丸混合粉的高效液相指纹图谱,从不同侧面突出反映不同类别化学成分对十全大补丸混合粉指纹图谱的系统贡献,从而实现对十全大补丸混合粉全方化学成分的检测,较全面的反映十全大补丸混合粉中各成分的现状,为整体控制和评价十全大补丸混合粉质量提供参考依据。
5.为实现上述目的及其他相关目的,本发明第一方面提供一种十全大补丸混合粉指纹图谱的检测方法,包括以下步骤:
6.1)供试品溶液的制备:将十全大补丸混合粉样品加入第一溶剂溶解,超声提取后冷却、摇匀、过滤,取续滤液即得供试品溶液;
7.2)对照品溶液的制备:将5-羟甲基糠醛、芍药苷、甘草苷、阿魏酸、1,2,3,4,6-o-五没食子酰葡萄糖、桂皮醛、甘草酸、藁本内酯对照品,加入第二溶剂溶解并定容,配成对照品溶液;
8.3)测定:采用相同色谱条件的高效液相色谱(hplc)法分别测定供试品溶液和对照品溶液,获得供试品溶液的指纹图谱和对照品溶液的指纹图谱,将供试品溶液的指纹图谱与对照品溶液的指纹图谱进行比较,对供试品溶液指纹图谱中的指标成分进行归属定位,从而获得十全大补丸混合粉指纹图谱。
9.优选地,步骤1)中,所述十全大补丸混合粉呈粉末状。所述十全大补丸混合粉是十
全大补丸生产加工过程中的中间体。
10.优选地,步骤1)中,所述十全大补丸混合粉样品加入的重量(g)与所述第一溶剂加入的体积(ml)之比为1:15-25。
11.更优选地,所述十全大补丸混合粉样品加入的重量(g)与所述第一溶剂加入的体积(ml)之比为1:20。
12.优选地,步骤1)中,所述第一溶剂为含1-3
‰
甲酸的65-75%甲醇。更优选地,所述第一溶剂为含2
‰
甲酸的70%甲醇。
13.所述65-75%甲醇为体积百分比为65-75%的甲醇水溶液。所述70%甲醇为体积百分比为70%的甲醇水溶液。所述1-3
‰
甲酸为体积百分比为1-3
‰
的甲酸。所述2
‰
甲酸为体积百分比为2
‰
的甲酸。
14.优选地,步骤1)中,所述十全大补丸混合粉样品加入第一溶剂后需要进行精密称重。
15.优选地,步骤1)中,所述超声提取时间为30-60min。更优选地,所述超声提取时间为45min。
16.优选地,步骤1)中,所述超声提取的功率为200-300w,所述超声提取的频率为30-50khz。更优选地,所述超声提取的功率为250w,所述超声提取的频率为40khz。
17.优选地,步骤1)中,所述冷却后需要再称重,并用第三溶剂补足失重。
18.更优选地,所述第三溶剂为65-75%甲醇。进一步优选地,所述第三溶剂为70%甲醇。
19.优选地,步骤1)中,所述摇匀后需要静置片刻。
20.更优选地,所述静置片刻以使超声提取后的溶液分层。
21.优选地,步骤1)中,所述过滤为取上清液过滤膜,放弃初滤液后,取续滤液。
22.更优选地,所述滤膜为0.45μm滤膜。
23.优选地,步骤2)中,所述第二溶剂为76-84%甲醇。更优选地,所述第二溶剂为80%甲醇。所述76-84%甲醇为体积百分比为76-84%的甲醇水溶液。所述80%甲醇为体积百分比为80%的甲醇水溶液。
24.优选地,步骤2)中,所述对照品溶液要摇匀、滤过,取续滤液。
25.优选地,步骤2)中,所述对照品溶液采用逐级稀释制得。
26.更优选地,所述逐级稀释采用的对照品储备溶液中,5-羟甲基糠醛的含量范围为39.27μg/ml,芍药苷的含量范围为637.46μg/ml,甘草苷的含量范围为88.71μg/ml,阿魏酸的含量范围为38.30μg/ml,1,2,3,4,6-o-五没食子酰葡萄糖的含量范围为84.27μg/ml,桂皮醛的含量范围为234.14μg/ml,甘草酸的含量范围为213.85μg/ml,藁本内酯的含量范围为440.80μg/ml。
27.优选地,步骤2)中,所述5-羟甲基糠醛的cas号为67-47-0,所述芍药苷的cas号为23180-57-6,所述甘草苷的cas号为551-15-5,所述阿魏酸的cas号为1135-24-6,所述1,2,3,4,6-o-五没食子酰葡萄糖的cas号为14937-32-7,所述桂皮醛的cas号为104-55-2,所述甘草酸的cas号为1405-86-3,所述藁本内酯的cas号为4431-01-0。
28.优选地,步骤2)中,所述对照品溶液中5-羟甲基糠醛的含量范围为0.004~0.196μg/ml,芍药苷的含量范围为0.064~3.187μg/ml,甘草苷的含量范围为0.009~0.444μg/ml,
阿魏酸的含量范围为0.004~0.192μg/ml,1,2,3,4,6-o-五没食子酰葡萄糖的含量范围为0.008~0.421μg/ml,桂皮醛的含量范围为0.023~1.171μg/ml,甘草酸的含量范围为0.021~1.069μg/ml,藁本内酯的含量范围为0.044~2.204μg/ml。
29.本技术中,选取5-羟甲基糠醛作为熟地黄的药效指标成分,阿魏酸、藁本内酯作为当归和川芎的药效指标成分,芍药苷和1,2,3,4,6-o-五没食子酰葡萄糖作为酒白芍的药效指标成分,桂皮醛作为肉桂的药效指标成分,甘草苷和甘草酸作为炙甘草的药效指标成分。
30.优选地,步骤3)中,所述供试品溶液的指纹图谱与对照品溶液的指纹图谱进行比较,是根据对照品溶液的指纹图谱中的已知特征峰,通过相对保留时间,指认出供试品溶液的指纹图谱中的相应特征峰,从而对供试品溶液指纹图谱中的指标成分进行归属定位。
31.优选地,步骤3)中,所述高效液相色谱法中的色谱柱为ods色谱柱。更优选地,所述高效液相色谱法中的色谱柱为dikma platisil ods色谱柱(4.6mm
×
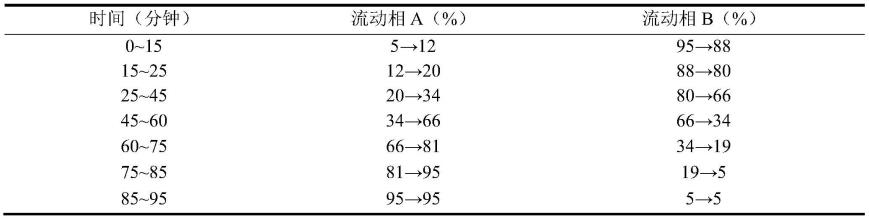
250mm,5μm)。
32.优选地,步骤3)中,所述高效液相色谱法中的检测器为光电二极管阵列检测器(dad)。
33.优选地,步骤3)中,所述高效液相色谱法中,当保留时间为0-36min时,柱温为15-25℃,优选为20℃;当保留时间为36-95min时,柱温为28-40℃,优选为30-35℃。
34.优选地,步骤3)中,所述高效液相色谱法中的进样量为1-10μl。更优选地,所述高效液相色谱法中的进样量为5μl。
35.优选地,步骤3)中,所述高效液相色谱法中,流速为0.8-1.2ml/min。更优选地,所述高效液相色谱法中,流速为1.0ml/min。
36.优选地,步骤3)中,所述高效液相色谱法中,采用波长切换的方式进行分析,其中,
37.当保留时间为0-7.5min时,检测波长为228-232nm,优选为230nm;
38.当保留时间为7.5-15min时,检测波长为263-267nm,优选为265nm;
39.当保留时间为15-25min时,检测波长为223-227nm,优选为225nm;
40.当保留时间为25-30min时,检测波长为241-243nm,优选为242nm;
41.当保留时间为30-44min时,检测波长为218-222nm,优选为220nm;
42.当保留时间为44-52min时,检测波长为239-241nm,优选为240nm;
43.当保留时间为52-63min时,检测波长为258-262nm,优选为260nm;
44.当保留时间为63-66min时,检测波长为288-292nm,优选为290nm;
45.当保留时间为66-95min时,检测波长为208-212nm,优选为210nm。
46.优选地,步骤3)中,所述高效液相色谱法中,流动相为乙腈-0.05-0.2%磷酸水溶液,其中,a相为乙腈,b相为0.05-0.2%磷酸水溶液;分析时间为95min;梯度洗脱。
47.更优选地,所述高效液相色谱法中,流动相为乙腈-0.1%磷酸水溶液,其中,a相为乙腈,b相为0.1%磷酸水溶液;分析时间为95min;梯度洗脱。
48.所述0.05-0.2%磷酸水溶液为体积百分数为0.05-0.2%的磷酸水溶液。所述0.1%磷酸水溶液为体积百分数为0.1%的磷酸水溶液。
49.更优选地,如表1所示,所述梯度洗脱的具体程序为:
50.0-15min,a相:b相体积比为5:95-12:88;
51.15-25min,a相:b相体积比为12:88-20:80;
52.25-45min,a相:b相体积比为20:80-34:66;
53.45-60min,a相:b相体积比为34:66-66:34;
54.60-75min,a相:b相体积比为66:34-81:19;
55.75-85min,a相:b相体积比为81:19-95:5;
56.85-95min,a相:b相体积比为95:5-95:5。
57.表1梯度洗脱程序
[0058][0059]
本发明第二方面提供一种十全大补丸混合粉指纹图谱的检测方法在十全大补丸混合粉中成分的质量检测中的用途。
[0060]
本发明第三方面提供一种十全大补丸混合粉的质量检测方法,包括采用前述十全大补丸混合粉指纹图谱的检测方法获得十全大补丸混合粉指纹图谱,将得到的十全大补丸混合粉指纹图谱与相同指纹图谱检测条件下获得的十全大补丸混合粉的对照指纹图谱进行相似度比较。
[0061]
优选地,本发明将测得的十全大补丸混合粉指纹图谱与十全大补丸混合粉的对照指纹图谱进行相似度的比较时,采用国家药典委员会发布的《中药色谱指纹图谱相似度评价系统》2012版软件进行比较。更优选地,本发明测得的十全大补丸混合粉指纹图谱与十全大补丸混合粉的对照指纹图谱的相似度≥0.9,优选为≥0.95。
[0062]
更优选地,所述十全大补丸混合粉指纹图谱与十全大补丸混合粉的对照指纹图谱进行指纹共有峰的匹配时,采用多点校正后在时间窗宽度为0.20min时进行自动全谱匹配,用平均数法生成指纹图谱和对照指纹图谱。
[0063]
优选地,采用与前述十全大补丸混合粉指纹图谱的检测方法相同的条件得到十全大补丸混合粉的对照指纹图谱,所述十全大补丸混合粉的对照指纹图谱包括30个共有指纹峰,以10号峰为参照峰(s峰,相对保留时间为1.0000),其他29个共有指纹峰的相对保留时间依次为1号峰(0.1485
±
0.0010)、2号峰(0.3349
±
0.0013)、3号峰(0.3998
±
0.0015)、4号峰(0.4284
±
0.0018)、5号峰(0.6467
±
0.0019)、6号峰(0.8151
±
0.0020)、7号峰(0.8300
±
0.0018)、8号峰(0.8450
±
0.0018)、9号峰(0.9443
±
0.0025)、11号峰(1.1390
±
0.0025)、12号峰(1.1660
±
0.0027)、13号峰(1.1914
±
0.0025)、14号峰(1.2132
±
0.0029)、15号峰(1.2319
±
0.0038)、16号峰(1.8618
±
0.0047)、17号峰(1.8876
±
0.0051)、18号峰(2.0645
±
0.0051)、19号峰(2.1562
±
0.0051)、20号峰(2.2370
±
0.0058)、21号峰(2.2832
±
0.0061)、22号峰(2.3282
±
0.0067)、23号峰(2.3476
±
0.0066)、24号峰(2.3833
±
0.0075)、25号峰(2.5833
±
0.0074)、26号峰(2.6956
±
0.0082)、27号峰(2.8284
±
0.0088)、28号峰(2.9415
±
0.0089)、29号峰(2.9781
±
0.0090)、30号峰(3.1807
±
0.0095)。
[0064]
所述十全大补丸混合粉的对照指纹图谱的具体数据见图1b。
[0065]
更优选地,所述十全大补丸混合粉的对照指纹图谱与对照品溶液的指纹图谱进行比较,定位确定所述3号峰为5-羟甲基糠醛的指纹峰,所述10号峰为芍药苷的指纹峰,所述
12号峰为甘草苷的指纹峰,所述14号峰为阿魏酸的指纹峰,所述15号峰为1,2,3,4,6-o-五没食子酰葡萄糖的指纹峰,所述16号峰为桂皮醛的指纹峰,所述17号峰为甘草酸的指纹峰,所述21号峰为藁本内酯的指纹峰。
[0066]
本发明第四方面提供一种十全大补丸混合粉中8种成分含量的测定方法,包括以下步骤:
[0067]
a)供试品溶液的制备:与十全大补丸混合粉指纹图谱的检测方法的步骤1)相同;
[0068]
b)对照品溶液的制备:与十全大补丸混合粉指纹图谱的检测方法的步骤2)相同;
[0069]
c)测定:采用与十全大补丸混合粉指纹图谱的检测方法中相同色谱条件的高效液相色谱法,分别测定步骤a)的供试品溶液和步骤b)的对照品溶液,并采用外标法计算供试品溶液中8种成分的含量。
[0070]
优选地,所述外标法是指:分别移取一系列不同体积的步骤b)所述对照品溶液,配成一系列不同浓度的溶液,采用高效液相色谱仪进样分析,获得对照品溶液中8种成分含量与峰面积的线性关系,以每一种成分色谱峰面积对应其相应的含量,绘制相应的标准工作曲线,计算得到各标准工作曲线的回归方程。再将供试品溶液采用高效液相色谱仪检测,将获得的供试品溶液中8种成分的色谱峰面积,分别代入所述各标准工作曲线的回归方程中,可得到相应成分的含量。
[0071]
更优选地,所述标准工作曲线中,以峰面积为纵坐标,以各对照品溶液的含量为横坐标。
[0072]
本发明第五方面提供一种十全大补丸混合粉中多味药材指纹图谱的筛选方法,包括下列步骤:
[0073]
a)单味药材样品溶液的制备:将十全大补丸混合粉中党参、炒白术、茯苓、炙甘草、当归、川芎、酒白芍、熟地黄、炙黄芪、肉桂10味药材样品中任一种或多种,按十全大补丸混合粉指纹图谱的检测方法的步骤1)进行制备,分别获得至少一种单味药材样品溶液;
[0074]
b)测定:采用与十全大补丸混合粉指纹图谱的检测方法的步骤3)中相同色谱条件的高效液相色谱(hplc)法测定单味药材样品溶液,获得单味药材样品溶液的指纹图谱;
[0075]
c)对照指纹图谱的取得:采用与十全大补丸混合粉指纹图谱的检测方法相同的步骤,得到十全大补丸混合粉的对照指纹图谱;
[0076]
d)质量检测:将单味药材样品溶液的指纹图谱,与十全大补丸混合粉的对照指纹图谱进行比较,通过相对保留时间,指认出单味药材样品溶液在十全大补丸混合粉的对照指纹图谱中的相应特征峰,从而对单味药材样品溶液的指纹图谱中的特征峰进行归属定位。
[0077]
优选地,步骤a)中,所述党参为桔梗科植物党参codonopsis pilosula(franch.)nannf.、素花党参codonopsis pilosula nannf.var.modesta(nannf.)l.t.shen或川党参codonopsis tangshen oliv.的干燥根。所述炒白术为白术的炒制加工品,为菊科植物白术atractylodes macrocephala koidz.的根茎。所述茯苓为多孔菌科真菌茯苓poria cocos(schw﹒)wolf.的干燥菌。所述炙甘草为甘草的炙制加工品,为豆科植物甘草glycyrrhiza uralensis fisch.、胀果甘草glycyrrhiza inflata bat.或光果甘草glycyrrhiza glabra l.的干燥根及根茎。所述当归为伞形科植物当归angelica sinensis(oliv.)diels.的根。所述川芎为伞形科植物川芎ligusticum chuanxiong hort.的根茎。所述酒白芍为甘草的
酒泡制加工品,为毛茛科植物芍药paeonia lactiflora pall.的根。所述熟地黄为玄参科植物地黄或rehmannia glutinosa libosch的根茎。所述炙黄芪为黄芪的炙制加工品,为豆科植物蒙古黄芪astragalus membranaceus fisch bge.var.mongholicus(bge.)hsiao和膜荚黄芪a.membranaceus(fisch.)bge.的根。所述肉桂为樟科植物肉桂cinnamomum cassia presl的干燥树皮。
[0078]
优选地,步骤d)中,本发明将测得的单味药材样品溶液的指纹图谱与十全大补丸混合粉的对照指纹图谱进行特征峰的归属定位,采用国家药典委员会发布的《中药色谱指纹图谱相似度评价系统》2012版软件进行分析处理,并通过十全大补丸混合粉的对照指纹图谱上各特征峰的相对保留时间对十全大补丸混合粉各单味药材的特征峰进行归属确证。具体结果见图3和表2。
[0079]
表2十全大补丸混合粉各单味药材的特征峰归属
[0080][0081]
优选地,步骤d)中,所述单味药材样品溶液中,党参样品溶液的指纹图谱包括1个共有指纹峰,所述1个共有指纹峰为5号峰。
[0082]
优选地,步骤d)中,所述单味药材样品溶液中,炒白术样品溶液的指纹图谱包括4个共有指纹峰,所述4个共有指纹峰为6号峰、22号峰、28号峰、30号峰。
[0083]
优选地,步骤d)中,所述单味药材样品溶液中,炙甘草样品溶液的指纹图谱包括6个共有指纹峰,所述6个共有指纹峰为7号峰、8号峰、11号峰、12号峰、22号峰、23号峰。
[0084]
优选地,步骤d)中,所述单味药材样品溶液中,当归样品溶液的指纹图谱包括11个共有指纹峰,所述11个共有指纹峰为5号峰、6号峰、14号峰、18号峰、20号峰、21号峰、24号峰、25号峰、26号峰、27号峰、29号峰。
[0085]
优选地,步骤d)中,所述单味药材样品溶液中,川芎样品溶液的指纹图谱包括5个共有指纹峰,所述5个共有指纹峰为6号峰、14号峰、18号峰、19号峰、21号峰。
[0086]
优选地,步骤d)中,所述单味药材样品溶液中,酒白芍样品溶液的指纹图谱包括6个共有指纹峰,所述6个共有指纹峰为2号峰、7号峰、9号峰、10号峰、13号峰、15号峰。
[0087]
优选地,步骤d)中,所述单味药材样品溶液中,熟地黄样品溶液的指纹图谱包括4个共有指纹峰,所述4个共有指纹峰为1号峰、3号峰、4号峰、27号峰。
[0088]
优选地,步骤d)中,所述单味药材样品溶液中,炙黄芪样品溶液的指纹图谱包括1个共有指纹峰,所述1个共有指纹峰为27号峰。
[0089]
优选地,步骤d)中,所述单味药材样品溶液中,肉桂样品溶液的指纹图谱包括1个共有指纹峰,所述1个共有指纹峰为16号峰。
[0090]
优选地,步骤d)中,所述茯苓样品溶液的指纹图谱不包括共有指纹峰。
[0091]
上述共有指纹峰中,确定3号峰为5-羟甲基糠醛的指纹峰,10号峰为芍药苷的指纹峰,12号峰为甘草苷的指纹峰,14号峰为阿魏酸的指纹峰,15号峰为1,2,3,4,6-o-五没食子酰葡萄糖的指纹峰,16号峰为桂皮醛的指纹峰,17号峰为甘草酸的指纹峰,21号峰为藁本内酯的指纹峰。
[0092]
本发明中的用水均为纯净水。
[0093]
如上所述,由于十全大补丸混合粉由十味中药材组成,所含化学成分极其复杂,本发明提供的一种十全大补丸混合粉指纹图谱及其成分的检测方法,建立十全大补丸混合粉的指纹图谱与多成分定量相结合的质量评价方法,定性定量分析两者优势互补。
[0094]
该种方法采用优化反应条件的前处理及高效液相色谱(hplc-dad)方法,建立了十全大补丸混合粉的高效液相指纹图谱,从不同侧面突出反映不同类别化学成分对十全大补丸混合粉指纹图谱系统的贡献,从而实现对十全大补丸混合粉全方化学成分的检测,能够较全面地反映十全大补丸混合粉的质量变化。
[0095]
本发明提供的方法还能够对十全大补丸混合粉中8个化学成分(5-羟甲基糠醛、芍药苷、1,2,3,4,6-o-五没食子酰葡萄糖、阿魏酸、藁本内酯、桂皮醛、甘草苷、甘草酸)进行多成分含量测定。该方法所测定的8种成分的标准曲线在各自范围内线性关系良好,重复性好,准确度高,精密度高。
[0096]
上述指纹图谱结合多成分同时定量方法,弥补了十全大补丸中间体检测方法的空白,具有简便、快速、准确的优点,重复性良好,可用于评价中间体质量一致性,为实现生产过程的全过程质量控制提供参考依据,以保证用药安全有效以及质量可控。
[0097]
上述方法还能够对十全大补丸混合粉中多个单味药材色谱峰进行了归属确证,指纹图谱体现了党参、炒白术、炙甘草、当归、川芎、酒白芍、熟地黄、炙黄芪、肉桂的特征峰。该方法可实现从原料源头进行有效监测,严格控制原药材的质量,并可用于生产过程的质量控制,从而保证临床用药的安全有效性。
附图说明
[0098]
图1显示为本发明中十全大补丸混合粉的对照指纹图谱及对照品的指纹图谱1a、1b,其中,图1a为对照品的指纹图谱,3:5-羟甲基糠醛,10:芍药苷,12:甘草苷,14:阿魏酸,15:1,2,3,4,6-o-五没食子酰葡萄糖,16:桂皮醛,17:甘草酸,21:藁本内酯;图1b为十全大补丸混合粉的对照指纹图谱。
[0099]
图2显示为本发明中多批次十全大补丸混合粉样品的叠加指纹图谱,其中,s1:200401;s2:200402;s3:200403;s4:200404;s5:200405;r(30):十全大补丸混合粉的对照指纹图谱。
[0100]
图3显示为本发明的十全大补丸混合粉中各特征峰归属图。
具体实施方式
[0101]
下面结合具体实施例进一步阐述本发明,应理解,这些实施例仅用于说明本发明而不用于限制本发明的保护范围。
[0102]
以下通过特定的具体实例说明本发明的实施方式,本领域技术人员可由本说明书所揭露的内容轻易地了解本发明的其他优点与功效。本发明还可以通过另外不同的具体实
施方式加以实施或应用,本说明书中的各项细节也可以基于不同观点与应用,在没有背离本发明的精神下进行各种修饰或改变。
[0103]
以下实施例使用的试剂和仪器如下:
[0104]
1、试剂
[0105]
对照品:阿魏酸(批号:10773-201915,质量分数99.4%)、甘草苷(批号:111610-201908,质量分数95.0%)均购自中国食品药品检定研究院;5-羟甲基糠醛(批号:7079,质量分数98.3%)、1,2,3,4,6-o-五没食子酰葡萄糖(批号:3367,质量分数98.0%)、芍药苷(批号:6461,质量分数96.3%)、桂皮醛(批号:5737,质量分数95.1%)、甘草酸(批号:5520,质量分数99.55%)均购自上海诗丹德标准技术服务有限公司;藁本内酯(批号:cx0036,质量分数》98%)购自江苏永健医药科技有限公司。
[0106]
样品:十全大补丸混合粉,由上海和黄药业有限公司提供,共5批次,(批号分别为200401、200402、200403、200404和200405)。、
[0107]
单味药材:黄芪、党参、熟地黄、白术、茯苓、当归、白芍、川芎、肉桂、炙甘草10味药材均购自安徽德昌药业股份有限公司,经打粉,过40目筛,贮存于密封袋中,备用。
[0108]
试剂:甲醇(分析纯ar,国药集团化学试剂有限公司),乙醇(分析纯ar,上海波尔化学试剂有限公司),乙腈(色谱纯,美国tedia公司),磷酸(色谱纯,美国tedia公司),超纯水由milli-q超纯水处理系统制备。
[0109]
2、仪器
[0110]
agilent 1260高效液相色谱仪(openlab cds 2.1色谱工作站,g1312b二元泵,g1322a自动脱气装置,g7116a柱温箱,g7115a dad检测器,g7129a自动进样器,美国agilent公司);sb-5200dtd型超声波清洗机(宁波新芝生物科技股份有限公司);电热鼓风干燥箱(上海一恒科学仪器有限公司);al204和xs205型分析电子天平(梅特勒-托利多mettler toledo仪器上海有限公司);milli-q advantage a10超纯水系统(德国默克密理博公司)。
[0111]
实施例1
[0112]
1、样品前处理
[0113]
供试品溶液的制备:取十全大补丸混合粉样品1.0g,精密称定,置于50ml的具塞锥形瓶中,精密加入含2
‰
甲酸的70%甲醇溶液20ml,密塞,称定重量,超声提取(功率250w,频率40khz)45分钟,放冷,再称定重量,用70%甲醇补足失重,摇匀,静置片刻,取上清液过0.45μm滤膜,取续滤液,即得供试品溶液1#。
[0114]
对照品溶液的制备:分别精密称取5-羟甲基糠醛、芍药苷、甘草苷、阿魏酸、1,2,3,4,6-o-五没食子酰葡萄糖、桂皮醛、甘草酸、藁本内酯对照品,加80%甲醇溶于同一100ml容量瓶中定容,摇匀,配成对照品储备溶液。对照品储备溶液中,5-羟甲基糠醛的含量范围为39.27μg/ml,芍药苷的含量范围为637.46μg/ml,甘草苷的含量范围为88.71μg/ml,阿魏酸的含量范围为38.30μg/ml,1,2,3,4,6-o-五没食子酰葡萄糖的含量范围为84.27μg/ml,桂皮醛的含量范围为234.14μg/ml,甘草酸的含量范围为213.85μg/ml,藁本内酯的含量范围为440.80μg/ml。
[0115]
再将对照品储备溶液精密量取,采用80%甲醇逐级稀释并定容,配成一系列不同浓度的对照品溶液。一系列不同浓度的对照品溶液中,5-羟甲基糠醛的含量范围为0.004~0.196μg/ml,芍药苷的含量范围为0.064~3.187μg/ml,甘草苷的含量范围为0.009~0.444
μg/ml,阿魏酸的含量范围为0.004~0.192μg/ml,1,2,3,4,6-o-五没食子酰葡萄糖的含量范围为0.008~0.421μg/ml,桂皮醛的含量范围为0.023~1.171μg/ml,甘草酸的含量范围为0.021~1.069μg/ml,藁本内酯的含量范围为0.044~2.204μg/ml。
[0116]
2、色谱条件
[0117]
所述高效液相色谱法的色谱条件为:色谱柱为dikma platisil ods色谱柱(4.6mm
×
250mm,5μm);检测器为光电二极管阵列检测器(dad);进样量:5μl;流速为1.0ml/min。
[0118]
当保留时间为0-36min时,柱温为20℃;当保留时间为36-95min时,柱温为33℃。
[0119]
采用波长切换的方式进行分析,其中,
[0120]
当保留时间为0-7.5min时,检测波长为230nm;
[0121]
当保留时间为7.5-15min时,检测波长为265nm;
[0122]
当保留时间为15-25min时,检测波长为225nm;
[0123]
当保留时间为25-30min时,检测波长为242nm;
[0124]
当保留时间为30-44min时,检测波长为220nm;
[0125]
当保留时间为44-52min时,检测波长为240nm;
[0126]
当保留时间为52-63min时,检测波长为260nm;
[0127]
当保留时间为63-66min时,检测波长为290nm;
[0128]
当保留时间为66-95min时,检测波长为210nm。
[0129]
流动相为乙腈-0.1%磷酸水溶液,其中,a相为乙腈,b相为0.1%磷酸水溶液;分析时间为95min;梯度洗脱。
[0130]
如表1所示,所述梯度洗脱的具体程序为:
[0131]
0-15min,a相:b相体积比为5:95-12:88;
[0132]
15-25min,a相:b相体积比为12:88-20:80;
[0133]
25-45min,a相:b相体积比为20:80-34:66;
[0134]
45-60min,a相:b相体积比为34:66-66:34;
[0135]
60-75min,a相:b相体积比为66:34-81:19;
[0136]
75-85min,a相:b相体积比为81:19-95:5;
[0137]
85-95min,a相:b相体积比为95:5-95:5。
[0138]
3、测定
[0139]
采用上述步骤2中的色谱条件的高效液相色谱法分别测定上述步骤1中供试品溶液1#和对照品溶液,获得供试品溶液1#的指纹图谱和对照品溶液的指纹图谱。其中,所述供试品溶液1#的指纹图谱,要与对照品溶液的指纹图谱进行比较,根据对照品溶液的指纹图谱中的已知特征峰,通过相对保留时间,指认出供试品溶液1#的指纹图谱中的相应特征峰,从而对供试品溶液1#指纹图谱中的指标成分进行归属定位,获得十全大补丸混合粉指纹图谱。
[0140]
实施例2
[0141]
1、样品前处理
[0142]
供试品溶液的制备:取十全大补丸混合粉样品1.0g,精密称定,置于50ml的具塞锥形瓶中,精密加入含1
‰
甲酸的65%甲醇溶液15ml,密塞,称定重量,超声提取(功率240w,频率30khz)50分钟,放冷,再称定重量,用65%甲醇补足失重,摇匀,静置片刻,取上清液过
0.45μm滤膜,取续滤液,即得供试品溶液2#。
[0143]
对照品溶液的制备:分别精密称取5-羟甲基糠醛、芍药苷、甘草苷、阿魏酸、1,2,3,4,6-o-五没食子酰葡萄糖、桂皮醛、甘草酸、藁本内酯对照品,加76%甲醇溶于同一100ml容量瓶中定容,摇匀,配成对照品储备溶液。再将对照品储备溶液精密量取,采用76%甲醇逐级稀释并定容,配成一系列不同浓度的对照品溶液。
[0144]
对照品储备溶液和一系列不同浓度的对照品溶液中,5-羟甲基糠醛、芍药苷、甘草苷、阿魏酸、1,2,3,4,6-o-五没食子酰葡萄糖、桂皮醛、甘草酸、藁本内酯的浓度范围同实施例1中步骤1。
[0145]
2、色谱条件
[0146]
所述高效液相色谱法的色谱条件为:色谱柱为dikma platisil ods色谱柱(4.6mm
×
250mm,5μm);检测器为光电二极管阵列检测器(dad);进样量:4μl;流速为0.8ml/min。
[0147]
当保留时间为0-36min时,柱温为20℃;当保留时间为36-95min时,柱温为30℃。
[0148]
采用波长切换的方式进行分析,其中,
[0149]
当保留时间为0-7.5min时,检测波长为231nm;
[0150]
当保留时间为7.5-15min时,检测波长为266nm;
[0151]
当保留时间为15-25min时,检测波长为226nm;
[0152]
当保留时间为25-30min时,检测波长为243nm;
[0153]
当保留时间为30-44min时,检测波长为221nm;
[0154]
当保留时间为44-52min时,检测波长为241nm;
[0155]
当保留时间为52-63min时,检测波长为261nm;
[0156]
当保留时间为63-66min时,检测波长为291nm;
[0157]
当保留时间为66-95min时,检测波长为211nm。
[0158]
流动相为乙腈-0.05%磷酸水溶液,其中,a相为乙腈,b相为0.05%磷酸水溶液;分析时间为95min;梯度洗脱。
[0159]
梯度洗脱的具体程序同实施例1中步骤2。
[0160]
3、测定
[0161]
具体测定过程同实施例1中步骤3。
[0162]
实施例3
[0163]
1、样品前处理
[0164]
供试品溶液的制备:取十全大补丸混合粉样品1.0g,精密称定,置于50ml的具塞锥形瓶中,精密加入含3
‰
甲酸的75%甲醇溶液25ml,密塞,称定重量,超声提取(功率260w,频率50khz)40分钟,放冷,再称定重量,用75%甲醇补足失重,摇匀,静置片刻,取上清液过0.45μm滤膜,取续滤液,即得供试品溶液3#。
[0165]
对照品溶液的制备:分别精密称取5-羟甲基糠醛、芍药苷、甘草苷、阿魏酸、1,2,3,4,6-o-五没食子酰葡萄糖、桂皮醛、甘草酸、藁本内酯对照品,加84%甲醇溶于同一100ml容量瓶中定容,摇匀,配成对照品储备溶液。再将对照品储备溶液精密量取,采用84%甲醇逐级稀释并定容,配成一系列不同浓度的对照品溶液。
[0166]
对照品储备溶液和一系列不同浓度的对照品溶液中,5-羟甲基糠醛、芍药苷、甘草苷、阿魏酸、1,2,3,4,6-o-五没食子酰葡萄糖、桂皮醛、甘草酸、藁本内酯的浓度范围同实施
例1中步骤1。
[0167]
2、色谱条件
[0168]
所述高效液相色谱法的色谱条件为:色谱柱为dikma platisil ods色谱柱(4.6mm
×
250mm,5μm);检测器为光电二极管阵列检测器(dad);进样量:6μl;流速为1.2ml/min。
[0169]
当保留时间为0-36min时,柱温为20℃;当保留时间为36-95min时,柱温为35℃。
[0170]
采用波长切换的方式进行分析,其中,
[0171]
当保留时间为0-7.5min时,检测波长为229nm;
[0172]
当保留时间为7.5-15min时,检测波长为264nm;
[0173]
当保留时间为15-25min时,检测波长为224nm;
[0174]
当保留时间为25-30min时,检测波长为241nm;
[0175]
当保留时间为30-44min时,检测波长为219nm;
[0176]
当保留时间为44-52min时,检测波长为239nm;
[0177]
当保留时间为52-63min时,检测波长为259nm;
[0178]
当保留时间为63-66min时,检测波长为289nm;
[0179]
当保留时间为66-95min时,检测波长为209nm。
[0180]
流动相为乙腈-0.2%磷酸水溶液,其中,a相为乙腈,b相为0.2%磷酸水溶液;分析时间为95min;梯度洗脱。
[0181]
梯度洗脱的具体程序同实施例1中步骤2。
[0182]
3、测定
[0183]
具体测定过程同实施例1中步骤3。
[0184]
实施例4
[0185]
采用上述实施例1中建立的十全大补丸混合粉指纹图谱的检测方法对5批次十全大补丸混合粉进行检测,获得供试品溶液的指纹图谱和对照品溶液的指纹图谱,得到的供试品的指纹图谱数据导入国家药典委员会发布的《中药色谱指纹图谱相似度评价系统》2012版软件中,以样品200401为参照图谱,采用多点校正后进行自动全谱匹配(时间窗为0.2min),用平均数法生成指纹图谱和对照指纹图谱,将供试品的指纹图谱与相同指纹图谱检测条件下获得的十全大补丸混合粉的对照指纹图谱进行相似度比较,并计算每批十全大补丸混合粉指纹图谱的相似度,结果5批十全大补丸混合粉指纹图谱的相似度均大于0.9,表明5批次样品的指纹图谱相似度较高(均≥0.95),总体质量较稳定,具体相似度情况见图2、表3。
[0186]
表3
[0187][0188]
实施例5
[0189]
采用上述实施例1中建立的十全大补丸混合粉指纹图谱的检测方法对十全大补丸混合粉进行检测,根据获得供试品溶液的指纹图谱和对照品溶液的指纹图谱,鉴定了30个
共有特征峰。
[0190]
具体的十全大补丸混合粉的对照指纹图谱见图1b,由图1b可知,根据色谱图中各色谱峰的相对保留时间,确定共有峰,其中hplc特征图谱的30个共有峰占总峰面积的比例达到90%以上,选取这30个共有峰作为特征指纹峰,具体数据见表4。其中10号峰出峰时间适中,分离度良好,峰面积较大,故以10号峰为参照峰(s峰,相对保留时间为1.0000),其他29个共有指纹峰的相对保留时间依次为1号峰(0.1485
±
0.0010)、2号峰(0.3349
±
0.0013)、3号峰(0.3998
±
0.0015)、4号峰(0.4284
±
0.0018)、5号峰(0.6467
±
0.0019)、6号峰(0.8151
±
0.0020)、7号峰(0.8300
±
0.0018)、8号峰(0.8450
±
0.0018)、9号峰(0.9443
±
0.0025)、11号峰(1.1390
±
0.0025)、12号峰(1.1660
±
0.0027)、13号峰(1.1914
±
0.0025)、14号峰(1.2132
±
0.0029)、15号峰(1.2319
±
0.0038)、16号峰(1.8618
±
0.0047)、17号峰(1.8876
±
0.0051)、18号峰(2.0645
±
0.0051)、19号峰(2.1562
±
0.0051)、20号峰(2.2370
±
0.0058)、21号峰(2.2832
±
0.0061)、22号峰(2.3282
±
0.0067)、23号峰(2.3476
±
0.0066)、24号峰(2.3833
±
0.0075)、25号峰(2.5833
±
0.0074)、26号峰(2.6956
±
0.0082)、27号峰(2.8284
±
0.0088)、28号峰(2.9415
±
0.0089)、29号峰(2.9781
±
0.0090)、30号峰(3.1807
±
0.0095)。
[0191]
表4指纹图谱共有峰相对保留时间n=6,x
±
sd
[0192][0193]
注:10号峰为定位峰(s峰)
[0194]
将上述的十全大补丸混合粉的对照指纹图谱,与对照品溶液的指纹图谱进行比较,根据图1a中的对照品溶液的指纹图谱中的已知特征峰,通过相对保留时间,指认出十全大补丸混合粉的对照指纹图谱中的相应特征峰,定位确定所述3号峰为5-羟甲基糠醛的指纹峰,所述10号峰为芍药苷的指纹峰,所述12号峰为甘草苷的指纹峰,所述14号峰为阿魏酸的指纹峰,所述15号峰为1,2,3,4,6-o-五没食子酰葡萄糖的指纹峰,所述16号峰为桂皮醛的指纹峰,所述17号峰为甘草酸的指纹峰,所述21号峰为藁本内酯的指纹峰。
[0195]
实施例6
[0196]
对本发明中的十全大补丸混合粉指纹图谱的检测方法进行方法学验证,其性能指标结果如下。
[0197]
1、精密度
[0198]
取200401批次的十全大补丸混合粉样品1份,按上述实施例1中十全大补丸混合粉
指纹图谱的检测方法进行检测,连续进样6次,记录色谱图,以芍药苷为参照峰,计算各共有峰相对保留时间的rsd《1.0%,相对峰面积的rsd《5.0%,表明仪器精密度良好。
[0199]
2、重复性
[0200]
取200401批次的十全大补丸混合粉样品6份,按上述实施例1中十全大补丸混合粉指纹图谱的检测方法进行检测,记录色谱图,以芍药苷为参照峰,计算各共有峰相对保留时间的rsd《1.0%,相对峰面积的rsd《5.0%,结果表明方法重复性良好。
[0201]
3、稳定性
[0202]
取200401批次的十全大补丸混合粉样品1份,按上述实施例1中十全大补丸混合粉指纹图谱的检测方法,制备供试品溶液后分别放置0h、2h、4h、8h、12h、24h进行检测,记录色谱图,以芍药苷为参照峰,计算各共有峰相对保留时间的rsd《1.0%,相对峰面积的rsd《5.0%,结果表明供试品溶液在24h内稳定性良好。
[0203]
实施例7
[0204]
1、样品前处理
[0205]
供试品溶液的制备:与实施例1中的步骤1中供试品溶液的制备过程相同。
[0206]
对照品溶液的制备:与实施例1中的步骤1中对照品溶液的制备过程相同。
[0207]
2、色谱条件
[0208]
所述高效液相色谱法的色谱条件与实施例1中的步骤2中高效液相色谱法的色谱条件相同。
[0209]
3、测定
[0210]
采用外标法,分别移取一系列不同体积的对照品溶液,配成一系列不同浓度的溶液,采用高效液相色谱仪进样分析,绘制标准工作曲线。再将获得的供试品溶液,采用高效液相色谱仪进样分析,并将分析结果代入标准工作曲线,即得供试品溶液中8种成分的含量。
[0211]
具体来说,分别移取一系列不同体积的对照品溶液,配成一系列不同浓度的溶液,采用高效液相色谱仪进样分析,获得对照品溶液中8种成分含量与峰面积的线性关系,以每一种成分色谱峰面积对应其相应的含量,绘制相应的标准工作曲线,计算得到各标准工作曲线的回归方程。再将供试品溶液采用高效液相色谱仪检测,将获得的供试品溶液中8种成分的色谱峰面积,分别代入所述各标准工作曲线的回归方程中,可得到相应成分的含量。
[0212]
实施例8
[0213]
配制上述8种对照品不同浓度的系列溶液,分别精密吸取对照品储备溶液0.5、1、2、5、10μl,采用实施例7的hplc检测条件,进行高效液相色谱仪分析,记录色谱图,以峰面积(y)为纵坐标,各对照品进样量(x)为横坐标,绘制标准曲线并进行线性回归计算,得到回归方程、相关系数和线性范围,具体结果见表5。
[0214]
由表5可知,回归方程的在相应浓度范围内进样时线性关系良好,相关系数r2》0.9999。
[0215]
表5工作曲线和检出限
[0216][0217]
实施例9
[0218]
1、精密度
[0219]
取200401批次的十全大补丸混合粉样品1份,按上述实施例7中方法进行检测,连续进样6次,记录峰面积,结果5-羟甲基糠醛、芍药苷、甘草苷、阿魏酸、1,2,3,4,6-o-五没食子酰葡萄糖、桂皮醛、甘草酸、藁本内酯峰面积的rsd均小于3.0%,表明仪器精密度良好。
[0220]
2、重复性
[0221]
取200401批次的十全大补丸混合粉样品6份,按上述实施例7中方法进行检测,记录峰面积,结果5-羟甲基糠醛、芍药苷、甘草苷、阿魏酸、1,2,3,4,6-o-五没食子酰葡萄糖、桂皮醛、甘草酸、藁本内酯峰面积的rsd均小于3.0%,表明方法重复性良好,准确度较高。
[0222]
3、稳定性
[0223]
取200401批次的十全大补丸混合粉样品1份,按上述实施例7中方法,制备供试品溶液后分别放置0h、2h、4h、8h、12h、24h进行检测,记录峰面积,结果5-羟甲基糠醛、芍药苷、甘草苷、阿魏酸、1,2,3,4,6-o-五没食子酰葡萄糖、桂皮醛、甘草酸、藁本内酯峰面积的rsd均小于3.0%,表明供试品溶液在24h内稳定性良好。
[0224]
4、加样回收率
[0225]
取批号为200401的十全大补丸混合粉样品9份,精密称定0.50g,按三个不同浓度水平分别加入5-羟甲基糠醛、芍药苷、甘草苷、阿魏酸、1,2,3,4,6-o-五没食子酰葡萄糖、桂皮醛、甘草酸、藁本内酯对照品适量,每一质量浓度取3份,按实施例7中步骤1制备获得供试品溶液,并按实施例7中步骤2的色谱条件,记录色谱图。计算8种有效成分在不同加入比例下的回收率,结果见表6,表明方法的准确度良好。
[0226]
表6加样回收试验结果(n=9)
[0227]
[0228][0229]
实施例10
[0230]
取5批十全大补丸混合粉样品(批号:200401、200402、200403、200404、200405),按实施例7中步骤1制备获得供试品溶液,并按实施例7中步骤2的色谱条件,分别进样分析,记录峰面积,采用外标法计算5-羟甲基糠醛、甘草苷、甘草酸、芍药苷、1,2,3,4,6-o-五没食子酰葡萄糖、阿魏酸、藁本内酯和桂皮醛含量,结果见表7。该方法能有效测定十全大补丸混合粉样品中8种成分的含量,方法操作简便、适用性好,结果准确、可靠。
[0231]
表7 5批十全大补丸混合粉中8个成分含量测定结果(mg
·
g-1
,n=5)
[0232][0233]
实施例11
[0234]
采用上述实施例1中样品前处理步骤,制备获得供试品溶液。
[0235]
分别取党参、炒白术、茯苓、炙甘草、当归、川芎、酒白芍、熟地黄、炙黄芪和肉桂10味药材粉末,采用上述实施例1中样品前处理步骤,制备获得10种单味药材样品溶液。
[0236]
采用实施例1的十全大补丸混合粉指纹图谱的检测方法中步骤2、3,分别测定供试品溶液、10种单味药材样品溶液,分别获得供试品溶液、10种单味药材样品溶液的指纹图谱。将得到的供试品溶液、10种单味药材样品溶液的指纹图谱导入国家药典委员会发布的《中药色谱指纹图谱相似度评价系统》2012版软件中进行分析处理,同时采用实施例1的十全大补丸混合粉指纹图谱的检测方法得到十全大补丸混合粉的对照指纹图谱。将供试品溶液、10种单味药材样品溶液的指纹图谱,分别与十全大补丸混合粉的对照指纹图谱进行比较,通过相对保留时间,指认出10种单味药材样品溶液在十全大补丸混合粉的对照指纹图谱中的相应特征峰,从而对10种单味药材样品溶液的指纹图谱中的特征峰进行归属定位,具体结果见图3。
[0237]
由图3可知,单味药材样品溶液中,党参、炒白术、茯苓、炙甘草、当归、川芎、酒白芍、熟地黄、炙黄芪和肉桂10味药材的样品溶液的指纹图谱中共有指纹峰的具体情况见表2。可见,十全大补丸混合粉中10味药材除茯苓外的化学特征峰在该指纹图谱中均有较好的体现,并得到归属确证。
[0238]
综上所述,本发明提供的一种十全大补丸混合粉指纹图谱的检测方法及其应用,建立了十全大补丸混合粉的hplc指纹图谱和多成分同时定量的分析方法,确定了30个共有特征峰,分析了共有峰来源,指认了8个成分,并对这8个化学成分(5-羟甲基糠醛、甘草苷、甘草酸、芍药苷、1,2,3,4,6-o-五没食子酰葡萄糖、阿魏酸、藁本内酯和桂皮醛)进行了定量分析。采用相似度分析对不同批次十全大补丸混合粉指纹图谱进行评价,同时对含量进行了分析,该方法稳定、可靠,精密度、重复性好,为更好地建立十全大补丸混合粉整体质量控制评价体系提供科学试验依据。所以,本发明克服了现有技术中的种种缺点而具高度产业利用价值。
[0239]
上述实施例仅例示性说明本发明的原理及其功效,而非用于限制本发明。任何熟悉此技术的人士皆可在不违背本发明的精神及范畴下,对上述实施例进行修饰或改变。因此,举凡所属技术领域中具有通常知识者在未脱离本发明所揭示的精神与技术思想下所完成的一切等效修饰或改变,仍应由本发明的权利要求所涵盖。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1