一种甲巯咪唑中有关物质的检测方法与流程
1.本技术涉及药品检测分析技术领域,特别是涉及一种甲巯咪唑中有关物质的检测方法。
背景技术:
2.甲状腺功能亢进症是当今人类内分泌系统疾病中的第二常见病。甲巯咪唑(thiamazole),化学名2-巯基-1-甲基咪唑,也叫他巴唑,为常用的抗甲状腺药物,临床上用于各种甲状腺功能亢进症(俗称“甲亢”)的治疗,特别适用于病情较轻,甲状腺轻中度肿大患者,其作用机制是抑制甲状腺素和三碘甲状腺原氨酸的合成,使免疫紊乱得到缓解。值得注意的是在该药物治疗过程中,由于甲巯咪唑的治疗窗口较窄,甲巯咪唑血药浓度过高时可能会导致患者出现恶心、头晕、腹泻甚至肾炎、红斑狼疮综合征和血小板减少等症状。
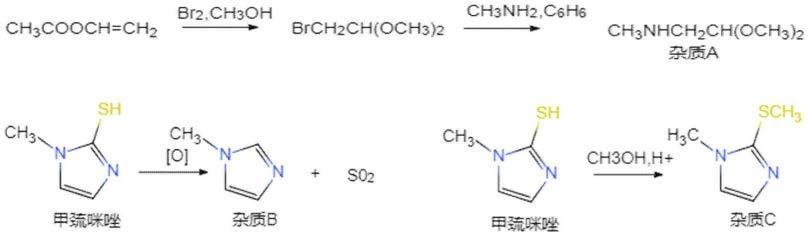
3.实际生产过程中,甲巯咪唑中不可避免的会含有多种杂质,根据合成路线判断,由于工业乙醇中常含有甲醇,因此按照反应路线会生成杂质a,在酸性条件下,杂质a中的缩醛结构有可能水解产生甲醇,甲醇进而攻击甲巯咪唑中的硫原子,发生脱水,生成杂质c,在氧化条件下,甲巯咪唑中的硫原子被氧化成二氧化硫即得到杂质b。这些杂质的存在会直接导致产品纯度低,影响药品的药性,且增加甲巯咪唑产品的安全风险,生成途径如下。
[0004][0005]
现有技术中,甲巯咪唑现行质量标准为中国药典,在中国药典中采用薄层色谱法对甲巯咪唑中的有关物质进行检测,由于薄层色谱法作为一种定性检查方法,检测的准确性较差,尚无法达到准确定量检测甲巯咪唑中有关物质含量的效果,且中国药典中规定总杂质限度为2%,仅对总杂质进行了控制,并没有对单个杂质进行限度要求,很难客观对甲巯咪唑的质量做出准确的评价。因此,研发一种对甲巯咪唑中有关物质的检测方法,以定量检测对甲巯咪唑中的杂质情况,从而准确全面控制甲巯咪唑中杂质的限度,进而提高甲巯咪唑药品安全性,对有效控制甲巯咪唑产品的纯度、药性和质量安全具有十分重要的意义。
技术实现要素:
[0006]
基于此,有必要针对现有技术中采用薄层色谱法对甲巯咪唑中的有关物质进行检测,检测的准确性较差,无法达到准确定量检测甲巯咪唑中有关物质含量的效果,且中国药典中规定总杂质限度为2%,仅对总杂质进行了控制,并没有对单个杂质进行限度要求,很难客观对甲巯咪唑的质量做出准确的评价。提供一种甲巯咪唑中有关物质的检测方法,利用气相色谱外标法-直接进样同时检测甲巯咪唑原料药中的杂质a、杂质b、杂质c、氨基乙醛
缩二乙醇和2-氯乙醛缩二乙醇,对现行质量标准中的方法进行修订,提高其检测的专属性、准确性和精密性从而准确全面的控制杂质限度,提高药品安全性,同时控制其它单个杂质和杂质总量从而更全面的控制各个药企原料药的杂质水平,客观对甲巯咪唑的质量做出准确的评价,为质量标准中有关物质检查项的制订提供参考和依据。
[0007]
一种甲巯咪唑中有关物质的检测方法,包括采用气相色谱外标法同时检测甲巯咪唑中的杂质a、杂质b、杂质c、氨基乙醛缩二乙醇和2-氯乙醛缩二乙醇,其包括以下步骤:
[0008]
s10.精密称定杂质a、杂质b、杂质c、氨基乙醛缩二乙醇和2-氯乙醛缩二乙醇,加乙醇溶解配制杂质a、杂质b、杂质c、氨基乙醛缩二乙醇质量浓度均为90μg/ml至110μg/ml、2-氯乙醛缩二乙醇质量浓度为2μg/ml至3μg/ml的混合对照品溶液;
[0009]
s20.精密称定甲巯咪唑,加乙醇溶解配制甲巯咪唑质量浓度为90mg/ml至110mg/ml的供试品溶液;
[0010]
s30.以直接进样方式将乙醇空白溶液、混合对照品溶液和供试品溶液分别注入气相色谱仪,记录色谱图,按外标法以峰面积计算甲巯咪唑中的杂质a、杂质b、杂质c、氨基乙醛缩二乙醇和2-氯乙醛缩二乙醇的含量。
[0011]
优选地,上述一种甲巯咪唑中有关物质的检测方法中,还包括采用气相色谱自身对照法检测甲巯咪唑中的其他杂质,其包括以下步骤:
[0012]
s40.精密量取供试品溶液,加乙醇溶解并稀释配制甲巯咪唑质量浓度为90μg/ml至110μg/ml的对照溶液;
[0013]
s50.以直接进样方式将对照溶液和供试品溶液分别注入气相色谱仪,记录色谱图,按自身对照法以峰面积计算甲巯咪唑中其他杂质的含量。
[0014]
优选地,上述一种甲巯咪唑中有关物质的检测方法中,对照溶液中甲巯咪唑的质量浓度为100μg/ml。
[0015]
优选地,上述一种甲巯咪唑中有关物质的检测方法中,色谱条件为:固定相为6%氰丙基苯基-94%二甲基聚硅氧烷的毛细管色谱柱;升温程序:起始柱温50℃至53℃,维持4分钟至5分钟,以每分钟9℃至11℃的速率升至97℃至105℃,保持6分钟至7分钟,再以每分钟28℃至32℃的速率升至248℃至253℃,保持10分钟至11分钟;氢火焰离子化检测器,温度为280℃至320℃;进样口温度为130℃至170℃,进样量为0.8μl至1.2μl,分流比为4:1至5:1;载气为氮气,流速为2.5ml/min至3.5ml/min。
[0016]
优选地,上述一种甲巯咪唑中有关物质的检测方法中,进样口温度为150℃,进样量为1μl,分流比为5:1。
[0017]
优选地,上述一种甲巯咪唑中有关物质的检测方法中,载气流速为3ml/min。
[0018]
优选地,上述一种甲巯咪唑中有关物质的检测方法中,氢火焰离子化检测器温度为300℃。
[0019]
优选地,上述一种甲巯咪唑中有关物质的检测方法中,色谱柱的规格为30m
×
0.53mm,3.0μm。
[0020]
优选地,上述一种甲巯咪唑中有关物质的检测方法中,升温程序:起始柱温50℃,维持5分钟,以每分钟10℃的速率升至100℃,保持6分钟,再以每分钟30℃的速率升至250℃,保持10分钟。
[0021]
优选地,上述一种甲巯咪唑中有关物质的检测方法中,所述供试品溶液中甲巯咪
唑的质量浓度为100mg/ml,混合对照品溶液中杂质a、杂质b、杂质c和氨基乙醛缩二乙醇的质量浓度均为100μg/ml、2-氯乙醛缩二乙醇的质量浓度2.5μg/ml。
[0022]
本技术采用的技术方案能够达到以下有益效果:
[0023]
本技术实施例公开的一种甲巯咪唑中有关物质的检测方法中,利用气相色谱外标法-直接进样同时检测甲巯咪唑原料药中的杂质a、杂质b、杂质c、氨基乙醛缩二乙醇和2-氯乙醛缩二乙醇,对现行质量标准中的方法进行修订,简便灵敏,检测适用性强、灵敏度高、专属性强,所得峰形对称,峰宽符合要求,各残留溶剂分离度好,检测结果准确可靠,适用于甲巯咪唑原料药中有关物质的常规检测,能够准确定量检测甲巯咪唑中有关物质含量,从而准确全面控制甲巯咪唑中杂质的限度,对单个杂质进行限度要求,进而提高甲巯咪唑药品安全性,有效控制甲巯咪唑产品的纯度、药性和质量安全,同时控制其它单个杂质和杂质总量从而更全面的控制各个药企原料药的杂质水平,客观对甲巯咪唑的质量做出准确的评价,为质量标准中有关物质检查项的制订提供参考和依据,进而全面控制甲巯咪唑药品质量使其符合用药安全要求,有效提高甲巯咪唑药品质量,降低临床用药风险。
附图说明
[0024]
图1为本技术三氯甲烷空白溶剂的气相色谱图;
[0025]
图2为本技术三氯甲烷溶解甲巯咪唑的气相色谱图;
[0026]
图3为本技术乙醇空白溶剂的气相色谱图;
[0027]
图4为本技术乙醇溶解甲巯咪唑的气相色谱图;
[0028]
图5为本技术甲巯咪唑系统适用性的气相色谱图;
[0029]
图6为本技术杂质a定位的气相色谱图;
[0030]
图7为本技术杂质b定位的气相色谱图;
[0031]
图8为本技术杂质c定位的气相色谱图;
[0032]
图9为本技术2-氯乙醛缩二乙醇定位的气相色谱图;
[0033]
图10为本技术氨基乙醛缩二乙醇定位的气相色谱图。
具体实施方式
[0034]
为了便于理解本技术,下面将参照相关附图对本技术进行更全面的描述。附图中给出了本技术的较佳实施方式。但是,本技术可以以许多不同的形式来实现,并不限于本文所描述的实施方式。相反地,提供这些实施方式的目的是使对本技术的公开内容理解的更加透彻全面。
[0035]
需要说明的是,当元件被称为“设置于”另一个元件,它可以直接在另一个元件上或者也可以存在居中的元件。当一个元件被认为是“连接”另一个元件,它可以是直接连接到另一个元件或者可能同时存在居中元件。本文所使用的术语“垂直的”、“水平的”、“左”、“右”、“顶部”、“底部”、“底端”、“顶端”以及类似的表述只是为了说明的目的,并不表示是唯一的实施方式。
[0036]
除非另有定义,本文所使用的所有的技术和科学术语与属于本技术的技术领域的技术人员通常理解的含义相同。本文中在本技术的说明书中所使用的术语只是为了描述具体的实施方式的目的,不是旨在于限制本技术。本文所使用的术语“和/或”包括一个或多个
相关的所列项目的任意的和所有的组合。
[0037]
本技术实施例公开一种甲巯咪唑中有关物质的检测方法,用于检测甲巯咪唑中的有关杂质,包括采用气相色谱外标法同时检测甲巯咪唑中的杂质a、杂质b、杂质c、氨基乙醛缩二乙醇和2-氯乙醛缩二乙醇,其包括以下步骤:
[0038]
s10.精密称定杂质a、杂质b、杂质c、氨基乙醛缩二乙醇和2-氯乙醛缩二乙醇,加乙醇溶解配制杂质a、杂质b、杂质c、氨基乙醛缩二乙醇质量浓度均为90μg/ml至110μg/ml、2-氯乙醛缩二乙醇质量浓度为2μg/ml至3μg/ml的混合对照品溶液;
[0039]
分别取杂质a、杂质b、杂质c、氨基乙醛缩二乙醇和2-氯乙醛缩二乙醇适量,精密称定,用乙醇定量稀释制成每1ml中分别含甲巯咪唑杂质a 90μg至110μg、杂质b 90μg至110μg、杂质c 90μg至110μg、氨基乙醛缩二乙醇90μg至110μg和2-氯乙醛缩二乙醇2μg至3μg的混合溶液。优选地,混合对照品溶液中杂质a、杂质b、杂质c和氨基乙醛缩二乙醇的质量浓度均为100μg/ml、2-氯乙醛缩二乙醇的质量浓度2.5μg/ml,即用乙醇定量稀释制成每1ml中分别含甲巯咪唑杂质a 0.1mg、杂质b 0.1mg、杂质c 0.1mg、氨基乙醛缩二乙醇0.1mg和2-氯乙醛缩二乙醇2.5μg的混合溶液。
[0040]
s20.精密称定甲巯咪唑,加乙醇溶解配制甲巯咪唑质量浓度为90mg/ml至110mg/ml的供试品溶液;
[0041]
精密称定甲巯咪唑0.9g至1.1g,置10ml量瓶中,加乙醇溶解并稀释至刻度,摇匀,便可以得到甲巯咪唑质量浓度为90mg/ml至110mg/ml的供试品溶液。优选地,供试品溶液中甲巯咪唑的质量浓度为100mg/ml,即精密称定甲巯咪唑1g,置10ml量瓶中,加乙醇溶解并稀释至刻度,摇匀即可。
[0042]
s30.以直接进样方式将乙醇空白溶液、混合对照品溶液和供试品溶液分别注入气相色谱仪,记录色谱图,按外标法以峰面积计算甲巯咪唑中的杂质a、杂质b、杂质c、氨基乙醛缩二乙醇和2-氯乙醛缩二乙醇的含量。
[0043]
按外标法,以峰面积计算的公式和计算过程均为已知技术,为了文本简洁,在此不再赘述。各成分峰均能达到基线分离,分离度大于1.5;溶剂对样品测定无干扰,杂质峰和主峰分离良好。
[0044]
根据本技术公开的检测方法和测定结果,在制定杂质种类控制时,对杂质a、杂质b、杂质c、氨基乙醛缩二乙醇和2-氯乙醛缩二乙醇5个已知杂质进行控制,按外标法以峰面积计算,杂质a、杂质b、杂质c均不得过0.1%,氨基乙醛缩二乙醇不得过0.1%,2-氯乙醛缩二乙醇不得过0.0025%。
[0045]
本技术实施例公开的一种甲巯咪唑中有关物质的检测方法中,利用气相色谱外标法-直接进样同时检测甲巯咪唑原料药中的杂质a、杂质b、杂质c、氨基乙醛缩二乙醇和2-氯乙醛缩二乙醇,对现行质量标准中的方法进行修订,简便灵敏,检测适用性强、灵敏度高、专属性强,所得峰形对称,峰宽符合要求,各残留溶剂分离度好,检测结果准确可靠,适用于甲巯咪唑原料药中有关物质的常规检测,能够准确定量检测甲巯咪唑中有关物质含量,从而准确全面控制甲巯咪唑中杂质的限度,对单个杂质进行限度要求,进而提高甲巯咪唑药品安全性,有效控制甲巯咪唑产品的纯度、药性和质量安全,同时控制其它单个杂质和杂质总量从而更全面的控制各个药企原料药的杂质水平,客观对甲巯咪唑的质量做出准确的评价,为质量标准中有关物质检查项的制订提供参考和依据,进而全面控制甲巯咪唑药品质
量使其符合用药安全要求,有效提高甲巯咪唑药品质量,降低临床用药风险。
[0046]
甲巯咪唑中除了上述的五种主要杂质外,还含有其他杂质,因此,还需要对其他杂质进行检测,更加准确全面控制甲巯咪唑中单个杂质和杂质总量的限度,有效控制甲巯咪唑产品的纯度、药性和质量安全。基于此,在一种可选的实施例中,本技术公开的检测方法还可以包括采用气相色谱自身对照法检测甲巯咪唑中的其他杂质,其包括以下步骤:
[0047]
s40.精密量取供试品溶液,加乙醇溶解并稀释配制甲巯咪唑质量浓度为90μg/ml至110μg/ml的对照溶液;
[0048]
精密量取供试品溶液1ml置100ml量瓶中,加乙醇溶解并稀释至刻度,精密量取上述溶液1ml置10ml量瓶中,加乙醇溶解并稀释至刻度,摇匀,便可以得到甲巯咪唑质量浓度为90μg/ml至110μg/ml的对照溶液。优选地,对照溶液中甲巯咪唑的质量浓度为100μg/ml。
[0049]
s50.以直接进样方式将对照溶液和供试品溶液分别注入气相色谱仪,记录色谱图,按自身对照法以峰面积计算甲巯咪唑中其他杂质的含量。
[0050]
按自身对照法,以峰面积计算的公式和计算过程均为已知技术,为了文本简洁,在此不再赘述。
[0051]
根据本技术公开的检测方法和测定结果,在制定其他杂质控制时,其他单个杂质不得过对照溶液主峰面积的1倍(即0.1%),杂质总量不得过对照溶液主峰面积的5倍(即0.5%)。
[0052]
通过气相色谱自身对照法检测甲巯咪唑中的其他杂质,对现行质量标准中的方法进行修订,简便灵敏,检测适用性强、灵敏度高、专属性强,所得峰形对称,峰宽符合要求,各残留溶剂分离度好,检测结果准确可靠,能够准确定量检测甲巯咪唑中有关物质含量,从而准确全面控制甲巯咪唑中其他杂质的限度,对其他未知杂质和杂质总量进行控制,进而提高甲巯咪唑药品安全性,有效控制甲巯咪唑产品的纯度、药性和质量安全,同时控制其它单个杂质和杂质总量从而更全面的控制各个药企原料药的杂质水平,客观对甲巯咪唑的质量做出准确的评价,为质量标准中有关物质检查项的制订提供参考和依据,进而全面控制甲巯咪唑药品质量使其符合用药安全要求,有效提高甲巯咪唑药品质量,降低临床用药风险。
[0053]
如上文所述,以直接进样方式将乙醇空白溶液、混合对照品溶液和供试品溶液分别注入气相色谱仪,或者,以直接进样方式将对照溶液和供试品溶液分别注入气相色谱仪,通过气相色谱仪检测过程中,需对气相色谱仪进行设置,设置其各项参数条件,可选地,在本技术中色谱条件可以为:采用固定相为6%氰丙基苯基-94%二甲基聚硅氧烷(或极性相近)的毛细管色谱柱,此种色谱柱分离性能较强。进一步地,色谱柱的规格可以为30m
×
0.53mm,3.0μm。
[0054]
杂质a沸点为140℃,杂质b沸点为198℃,杂质c沸点为166℃,2-氯乙醛缩二乙醇沸点为157.4℃,氨基乙醛缩二乙醇沸点163℃,为了使各个杂质之间分离度达到要求,设置升温程序:起始柱温50℃至53℃,维持4分钟至5分钟,以每分钟9℃至11℃的速率升至97℃至105℃,保持6分钟至7分钟,再以每分钟28℃至32℃的速率升至248℃至253℃,保持10分钟至11分钟,通过此种程序升温,使各个杂质之间分离度达到要求。具体地,各个杂质之间的分离度大于1.5。
[0055]
优选地,设置升温程序:起始柱温50℃,维持5分钟,以每分钟10℃的速率升至100℃,保持6分钟,再以每分钟30℃的速率升至250℃,保持10分钟。
[0056]
检测器可以选择氢火焰离子化检测器,检测器温度可以为280℃至320℃;具体地,氢火焰离子化检测器温度可以为300℃。
[0057]
进样口温度可以为130℃至170℃,进样量可以为0.8μl至1.2μl,分流比可以为4:1至5:1;具体地,进样口温度可以为150℃,进样量可以为1μl,分流比可以为5:1。
[0058]
可以选择载气为氮气,流速可以为2.5ml/min至3.5ml/min;具体地,载气流速可以为3ml/min。
[0059]
以下通过具体实验过程,进一步说明本技术的技术方案以及技术效果。
[0060]
本次研究目的为修订有关物质检查方法,提高其检测的专属性、准确性和精密性从而准确全面的控制杂质限度,提高药品安全性。具体方案为将原有薄层色谱法修订为专属性更优的气相色谱法,将甲巯咪唑中的杂质a、杂质b、杂质c、氨基乙醛缩二乙醇和2-氯乙醛缩二乙醇设置为特殊杂质,同时控制其它单个杂质和杂质总量从而更全面的控制各个药企原料药的杂质水平。根据杂质a、杂质b、杂质c、氨基乙醛缩二乙醇和2-氯乙醛缩二乙醇不同的性质设置色谱条件,对现行质量标准中的方法进行修订。
[0061]
(1)色谱柱和进样方式的选择
[0062]
本次研究方法考察采用分离性能较强的固定相为6%氰丙基苯基-94%二甲基聚硅氧烷的毛细管色谱柱(30m
×
0.53mm,3.0μm),进样方式采用直接进样。
[0063]
(2)柱温箱程序升温条件的选择
[0064]
杂质a沸点为140℃,杂质b沸点为198℃,杂质c沸点为166℃,2-氯乙醛缩二乙醇沸点为157.4℃,氨基乙醛缩二乙醇沸点163℃,为了使各个杂质之间分离度达到要求,设置程序升温条件为起始柱温50℃,维持5分钟,以每分钟10℃的速率升至100℃,维持6分钟,再以每分钟30℃的速率升至250℃,维持10分钟。
[0065]
(3)进样口与检测器温度选择
[0066]
设置进样口温度为150℃,fid检测器(氢火焰离子化检测器)温度为300℃。
[0067]
(4)溶剂的选择
[0068]
甲巯咪唑在乙醇和三氯甲烷中易溶,现有技术中采用三氯甲烷作溶剂,考虑到在破坏实验中酸、碱、氧化试剂均为水溶液,其与三氯甲烷不相溶,故选择乙醇作为溶剂进行考察,并与三氯甲烷做一对比。
[0069]
精密称取2份甲巯咪唑10mg,分别置10ml容量瓶,分别用三氯甲烷和乙醇溶解并稀释定容,取供试品溶液按照上述的有关物质色谱条件进样,色谱图见图1至图4。
[0070]
从图谱上可以得知:1、三氯甲烷空白溶剂中有9个色谱峰,乙醇空白溶剂中有5个色谱峰;2、三氯甲烷溶解样品后的供试品中可以检测出两个色谱峰,分别为保留时间20.782的未知杂质峰和保留时间24.701的甲巯咪唑峰。乙醇溶解样品后的供试品中可以检测出四个色谱峰,分别为保留时间21.399、22.331和23.115的未知杂质峰和保留时间24.706的甲巯咪唑峰。为了减小空白溶剂对供试品溶液色谱峰的影响及更大程度的使供试品中杂质溶出,及降低试剂对试验人员身体的伤害,故选择乙醇作为样品的溶剂。
[0071]
(5)供试品溶液浓度的选择
[0072]
将甲巯咪唑和5种杂质不断稀释,取稀释溶液按照上述的有关物质色谱条件进样,直至信噪比为3:1时为最低检出浓度,确定每种杂质的最低检出浓度,采用“上推法”即供试品浓度应是最低检出浓度的2000至5000倍,来确定供试品溶液浓度。甲巯咪唑和杂质的最
低检出浓度见表1。
[0073]
表1 甲巯咪唑及其有关物质最低检测浓度
[0074]
项目最低检测浓度(μg/ml)杂质a45.68杂质b3.34杂质c1.41氨基乙醛缩二乙醇18.162-氯乙醛缩二乙醇2.00甲巯咪唑1.01
[0075]
为了保证有足够的检测灵敏度,选择最低检测浓度最大值进行设定,同时考虑仪器和色谱柱承载的因素,设定对供试品溶液为最低检出浓度2000倍,即92mg/ml,考虑到称样的方便性与可操作性,最终设定供试品溶液为100mg/ml。
[0076]
(6)系统适用性试验
[0077]
购买杂质a、杂质b、杂质c、氨基乙醛缩二乙醇和2-氯乙醛缩二乙醇,配制成含各杂质均约为100μg/ml及甲巯咪唑100mg/ml的溶液,采用上述的有关物质色谱条件进行检查,色谱图见图5至图10。
[0078]
色谱图显示各杂质之间及各杂质与主峰之间均可以有效分离,拟将“各杂质100μg/ml及甲巯咪唑100mg/ml”作为系统适用性溶液配制浓度,系统适用性要求为各杂质之间及杂质与主峰间分离度应符合要求。
[0079]
(7)样品的测定
[0080]
按照上述检测方法,分别对北京市燕京、贵州圣济堂和merck kgaa公司提供的三批样品进行测定,结果见表2。
[0081]
表2 有关物质测定结果
[0082][0083]
检测结果显示在已知杂质检查方面:北京燕京检出已知杂质b、杂质c,merck kgaa检出已知杂质c;在其他未知单杂检查方面:merck kgaa公司检出的未知单杂数量最多,且三个厂家均检出保留时间约为6.8min和21min的杂质峰。
[0084]
(7)限度的确定
[0085]
根据样品用上述的检测方法测定结果,在制定杂质种类控制时,对杂质a、杂质b、杂质c、氨基乙醛缩二乙醇和2-氯乙醛缩二乙醇5个已知杂质进行控制,同时对其他未知杂质和杂质总量进行控制。杂质a、杂质b、杂质c的限度为:按外标法以峰面积计算,杂质a、杂质b、杂质c均不得过0.1%。杂质氨基乙醛缩二乙醇和2-氯乙醛缩二乙醇的限度为:按外标法以峰面积计算,氨基乙醛缩二乙醇不得过0.1%,2-氯乙醛缩二乙醇不得过0.0025%。按自身对照法以峰面积计算其他单个杂质不得过对照溶液主峰面积的1倍(0.1%),杂质总量
不得过对照溶液主峰面积的5倍(0.5%)。
[0086]
综上所述,利用气相色谱外标法-直接进样同时检测甲巯咪唑原料药中的杂质a、杂质b、杂质c、氨基乙醛缩二乙醇和2-氯乙醛缩二乙醇,通过气相色谱自身对照法检测甲巯咪唑中的其他杂质,对现行质量标准中的方法进行修订,简便灵敏,检测适用性强、灵敏度高、专属性强,所得峰形对称,峰宽符合要求,各残留溶剂分离度好,检测结果准确可靠,适用于甲巯咪唑原料药中有关物质的常规检测,能够准确定量检测甲巯咪唑中有关物质含量,从而准确全面控制甲巯咪唑中杂质的限度,对单个杂质进行限度要求,进而提高甲巯咪唑药品安全性,有效控制甲巯咪唑产品的纯度、药性和质量安全,同时控制其它单个杂质和杂质总量从而更全面的控制各个药企原料药的杂质水平,客观对甲巯咪唑的质量做出准确的评价,为质量标准中有关物质检查项的制订提供参考和依据,进而全面控制甲巯咪唑药品质量使其符合用药安全要求,有效提高甲巯咪唑药品质量,降低临床用药风险。
[0087]
以上所述实施例的各技术特征可以进行任意的组合,为使描述简洁,未对上述实施例中的各个技术特征所有可能的组合都进行描述,然而,只要这些技术特征的组合不存在矛盾,都应当认为是本说明书记载的范围。
[0088]
以上所述实施例仅表达了本技术的几种实施方式,其描述较为具体和详细,但并不能因此而理解为对申请专利范围的限制。应当指出的是,对于本领域的普通技术人员来说,在不脱离本技术构思的前提下,还可以做出若干变形和改进,这些都属于本技术的保护范围。因此,本技术专利的保护范围应以所附权利要求为准。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1