一种西黄胶囊的指纹图谱构建方法及指纹图谱
1.本发明属于复方中药制剂品控管理技术领域,具体涉及一种西黄胶囊的指纹图谱构建方法及指纹图谱。
背景技术:
2.西黄胶囊源于清代著名医家王洪绪《外科全生集》中古方西黄丸,经现代工艺由牛黄、麝香、乳香、没药四味中药精制而成。方中牛黄清心、退热化痰、通窍、散肿结为主药,辅以麝香芳香辛串之性,通经络,散瘀消肿;乳香与各药相互配合,活血化瘀消肿定痛,全方配合,清热解毒,活血祛瘀,消坚肿。目前临床上常用来治疗乳腺纤维瘤、乳癌、颈淋巴结结核、淋巴结炎、骨髓炎、阑尾炎、化脓性皮肤炎、多发性脓肿、菌血症、急性化脓性感染和恶性肿瘤等病症,经多年临床应用,疗效显著安全可靠,抗肿瘤谱广,是治疗肿瘤、组织增生性和感染性疾病的首选用药。
3.目前关于西黄胶囊的质量检测方法较少,多数为方中单味药的成分研究,不能反映西黄胶囊的整体成分信息,因此其质量无法很好的控制。
技术实现要素:
4.本发明的目的是为了解决现有技术的不足,以提供一种西黄胶囊的指纹图谱构建方法及指纹图谱,该方法可以准确、清晰、客观的评价西黄胶囊的质量,对于控制西黄胶囊的质量以及提高和保证临床疗效具有重要作用和价值。
5.本发明通过以下技术方案实现:
6.一种西黄胶囊的指纹图谱构建方法,包括以下步骤:
7.s1、取西黄胶囊内容物,加甲醇-氯仿-磷酸溶液,超声提取,得西黄胶囊供试品溶液;
8.s2、取胆酸、猪去氧胆酸、去氧胆酸、胆红素、麝香酮和没药酮,溶于乙醇,得混合标准品溶液1;取苦木素、11-羰基-β-乳香酸、11-羰基-β-乙酰乳香酸、乙酰-11α-甲氧基-β-乳香酸和山达海松酸,溶于甲醇,得混合标准品溶液2;
9.s3、将西黄胶囊供试品溶液、混合标准品溶液1和混合标准品溶液2分别注入高效液相色谱仪,进行色谱分析,记录相应的色谱图;
10.s4、根据s3中获得的西黄胶囊供试品溶液的色谱图及混合标准品溶液1 和混合标准品溶液2的色谱图,构建得到西黄胶囊的指纹图谱。
11.优选的,s1中,甲醇-氯仿-磷酸溶中,甲醇和氯仿的体积比为1:3,甲醇和氯仿的总量与磷酸的体积比为100:0.2。
12.优选的,s3中,高效液相色谱仪采用的色谱柱为hypersil c18 ods。
13.优选的,s3中,色谱分析采用的流动相为甲醇-0.08%磷酸-乙腈。
14.进一步的,s3中,色谱分析时采用紫外可见吸收检测器多波长切换检测,检测波长:0~15min:254nm,15~45min:249nm,45~90min:239nm, 90~110min:210nm。
15.进一步的,s3中,色谱分析时,梯度洗脱程序为:0~15min甲醇体积54%
→
65%,0.08%磷酸体积36%
→
25%;15~45min甲醇体积65%
→
78%,0.08%磷酸体积25%
→
12%;45~55min甲醇体积78%
→
80%,0.08%磷酸体积 12%
→
10%;55~65min甲醇体积80%
→
82%,0.08%磷酸体积10%
→
8%; 65~75min甲醇体积82%
→
84%,0.08%磷酸体积8%
→
6%;75~90min甲醇体积84%
→
86%,0.08%磷酸体积6%
→
4%;90~100min甲醇体积86%
→
88%, 0.08%磷酸体积4%
→
2%;100~110min甲醇体积88%
→
90%,0.08%磷酸体积2%
→
0%。
16.优选的,s4具体是:将不同批次的西黄胶囊供试品溶液的色谱图导入中药色谱指纹图谱相似度评价系统2004a,选择不同批次西黄胶囊的色谱图中均存在的色谱峰作为共有峰,用平均值计算法生成西黄胶囊的对照指纹图谱,计算各共有峰的相对保留时间和相对峰面积;并根据混合标准品溶液1和混合标准品溶液2的色谱图的保留时间标注对照指纹图谱中各共有峰的化学成分;采用对照指纹图谱r生成共有色谱峰模式,分析计算获得各批西黄胶囊的色谱图与共有色谱峰之间具的相似性,确认各批西黄胶囊供试品溶液的色谱图的可靠性;
17.将s3中获得的西黄胶囊供试品溶液的色谱图与混合标准品溶液1和混合标准品溶液2的色谱图进行比对,指认出色谱图中3号峰为没药酮;4号峰为山达海松酸;8号峰为苦木素;10号峰为麝香酮;11号峰为11-羰基-β-乳香酸; 12号峰为11-羰基-β-乙酰乳香酸;13号峰为乙酰-11α-甲氧基-β-乳香酸,得到西黄胶囊指纹图谱。
18.采用所述的方法构建得到的西黄胶囊指纹图谱。
19.与现有技术相比,本发明具有如下的有益效果:
20.中药指纹图谱可以整体、宏观地表征药品主要化学成分的特征,是目前公认的最适合中药材及中成药化学成分质量控制的手段之一,与现代分析技术相结合的中药指纹图谱,用能体现中药药效物质基础的化学物质群来综合评价中药质量,在中药质量标准制定中被广泛应用。基于此,本发明提供一种西黄胶囊指纹图谱的构建方法,本发明对不同提取方法和提取溶剂进行了实验考察,优化提取方法和溶剂,得到的色谱图信息量多,成分含量高,本发明提供的高效液相色谱指纹图谱检测方法系统适应性良好、专属性强、精密度高、稳定性好、可重复性强,符合构建指纹图谱的要求,有利于更全面、更有效地控制西黄胶囊临床用药的质量,使其安全性和疗效得到更好的保障,可应用于西黄胶囊的质量控制,对西黄胶囊的成分鉴别、质量评价以及质量标准的制定具有重要的意义。用本发明所提供的方法所建立的西黄胶囊的指纹图谱共计13个特征共有峰,并指认其中7个特征峰。能有效地表征西黄胶囊的质量,能客观体现各个构成指纹特征峰的前后顺序和相互关系,注重整体面貌特征,既可避免因测定个别化学成分而判定西黄胶囊质量的片面性,又可减少为质量达标而人为处理的可能性。
附图说明
21.图1为本发明混合标准品1色谱图。
22.图2为本发明混合标准品2色谱图。
23.图3为本发明西黄胶囊的11批次供试品指纹图谱。
24.图4为本发明没药酮对照品的质谱图。
25.图5为本发明山达海松酸对照品的质谱图。
26.图6为本发明苦木素对照品的质谱图。
27.图7为本发明麝香酮对照品的质谱图。
28.图8为本发明11-羰基-β-乳香酸对照品的质谱图。
29.图9为本发明11-羰基-β-乙酰乳香酸对照品的质谱图。
30.图10为本发明乙酰-11α-甲氧基-β-乳香酸对照品的质谱图。
具体实施方式
31.为了进一步理解本发明,下面结合实施例对本发明进行描述,这些描述只是进一步解释本发明的特征和优点,并非用于限制本发明的权利要求。
32.1.一种西黄胶囊的指纹图谱构建方法,包括以下步骤:
33.s1、西黄胶囊供试品溶液的制备:
34.分别精密称取不同批次的西黄胶囊内容物,置于具塞锥形瓶中,加甲醇
‑ꢀ
氯仿-磷酸溶液,超声提取,取滤液过0.45μm微孔滤膜,得西黄胶囊供试品溶液;
35.s2、标准品溶液的制备:
36.分别精密称定胆酸、猪去氧胆酸、去氧胆酸、胆红素、麝香酮、没药酮,置于容量瓶中,用乙醇定容至刻度,得混合标准品溶液1;精密称定苦木素、 11-羰基-β-乳香酸、11-羰基-β-乙酰乳香酸、乙酰-11α-甲氧基-β-乳香酸、山达海松酸,置于容量瓶中,用甲醇定容至刻度,摇匀,得混合标准品溶液2;
37.s3、分别精密吸取s1的供试品溶液和s2的混合标准品溶液1和混合标准品溶液2,注入高效液相色谱仪,记录色谱图;
38.s4、将s3中获得的西黄胶囊供试品溶液的色谱图导出,并导入中药色谱指纹图谱相似度评价系统2004a;选择不同批次西黄胶囊的色谱图中均存在的色谱峰作为共有峰,共有峰13个,用平均值计算法生成西黄胶囊的对照指纹图谱,计算各共有峰的相对保留时间和相对峰面积;并根据混合标准品溶液1 和混合标准品溶液2的色谱图的保留时间标注对照指纹图谱中峰的化学成分;采用对照指纹图谱r生成共有色谱峰模式,分析计算获得各批西黄胶囊的色谱图与共有色谱峰之间具的相似性,确认各批西黄胶囊供试品溶液的色谱图的可靠性;
39.s5、将s3中获得的西黄胶囊供试品溶液的色谱图和混合标准品溶液1和混合标准品溶液2的色谱图进行比对,指认出西黄胶囊图谱中3号峰为没药酮; 4号峰为山达海松酸;8号峰为苦木素;10号峰为麝香酮;11号峰为11-羰基-β
‑ꢀ
乳香酸;12号峰为11-羰基-β-乙酰乳香酸;13号峰为乙酰-11α-甲氧基-β-乳香酸,得到西黄胶囊的指纹图谱。
40.其中,s1中西黄胶囊供试品溶液制备方法,优选为:分别精密称取11批次的西黄胶囊内容物240mg,置于250ml具塞锥形瓶中,加[甲醇-氯仿(1:3)]
‑ꢀ
磷酸(100:0.2)溶液100ml,超声提取30min,滤液过0.45μm微孔滤膜,得供试品溶液。
[0041]
s2中标准品溶液的制备方法,优选为:分别精密称定胆酸、猪去氧胆酸、去氧胆酸、胆红素、麝香酮、没药酮,置于容量瓶中,用乙醇定容至刻度,得混合标准品溶液1;精密称定苦木素、11-羰基-β-乳香酸、11-羰基-β-乙酰乳香酸、乙酰-11α-甲氧基-β-乳香酸、山达海松酸,置于容量瓶中,用甲醇定容至刻度,摇匀,得混合标准品溶液2,制成的混合标准品溶
液中胆酸、猪去氧胆酸、去氧胆酸、胆红素、麝香酮、没药酮、苦木素、11-羰基-β-乳香酸、11-羰基-β
‑ꢀ
乙酰乳香酸、乙酰-11α-甲氧基-β-乳香酸、山达海松酸的浓度分别为200μg/ml、 200μg/ml、200μg/ml、40μg/ml、150μg/ml、150μg/ml、100μg/ml、100μg/ml、 100μg/ml、100μg/ml、150μg/ml。
[0042]
s3中液相色谱条件为:色谱柱:hypersil c18 ods(4.6
×
250mm
×
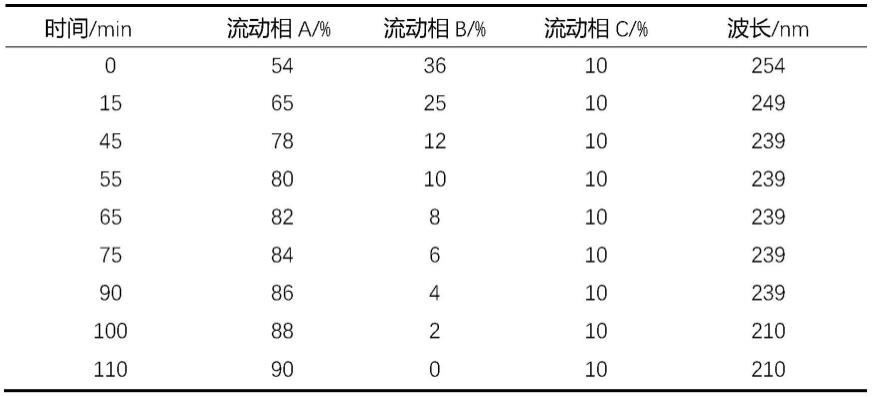
5μm);流动相:甲醇(a)-0.08%磷酸(b)-乙腈(c),梯度洗脱;紫外可见吸收检测器多波长切换检测,检测波长:0~15min:254nm、15~45min:249nm、 45~90min:239nm、90~110min:210nm;柱温:27℃;流速0.6ml/min;进样体积:20μl;洗脱程序如下表1。
[0043]
表1液相色谱洗脱程序
[0044][0045]
2.指纹图谱检测的优化过程:
[0046]
s1、在样品溶液制备方面的优化:
[0047]
本发明通过对不同提取方法(超声、回流、浸渍)进行实验考察,结果发现超声提取所得的谱图成分比较全面,分离度良好,故采用超声提取的方法;
[0048]
本发明比较了不同提取溶剂([乙腈-氯仿(1:3)]-磷酸(100:0.2)溶液、甲醇-氯仿(1:3)溶液、[甲醇-氯仿(1:3)]-磷酸(100:0.2)溶液)的提取效果进行比较,结果发现以[甲醇-氯仿(1:3)]-磷酸(100:0.2)溶液为提取溶剂时,提取物色谱图信息量最多,成分含量最高,所以选用[甲醇-氯仿(1: 3)]-磷酸(100:0.2)溶液进行提取;
[0049]
s2、在色谱条件方面进行优化:
[0050]
本发明采用紫外可见吸收检测器对检测波长进行考察,提取254nm、 249nm、239nm、210nm处的色谱图,发现检测波长条件为:0~15min,254 nm;15~45min,249nm;45~90min,239nm;90~110min,210nm时色谱图所包含的信息量最全面且基线平稳,故选此方法为检测波长条件。
[0051]
本发明对流速(0.6ml/min、0.8ml/min、1.0ml/min)进行筛选,发现流速为0.6ml/min时出峰情况最好,分离度最佳,故保持流速为0.6ml/min。
[0052]
本发明对柱温(25℃、27℃、30℃)进行筛选,结果发现,柱温保持27℃时出峰情况最佳,各成分分离效果较好,故最终选择柱温为27℃。
[0053]
本发明比较了甲醇-水、乙腈-水、甲醇-0.3%磷酸-水、乙腈-0.1%磷酸-水、甲醇-0.1%磷酸-乙腈、甲醇-0.08%磷酸-乙腈多个不同洗脱系统在不同梯度下的洗脱效果。结
果发现以甲醇-0.08%磷酸-乙腈为流动相时,西黄胶囊中各成分分离效果较好,故最终选定以甲醇-0.08%磷酸-乙腈为流动相。
[0054]
在确定最佳流动相组成后,本发明通过大量实验筛选最佳的梯度洗脱程序,实验发现,当采用0~15min甲醇体积54%
→
65%,0.08%磷酸体积36%
→
25%; 15~45min甲醇体积65%
→
78%,0.08%磷酸体积25%
→
12%;45~55min 甲醇体积78%
→
80%,0.08%磷酸体积12%
→
10%;55~65min甲醇体积80%
→
82%,0.08%磷酸体积10%
→
8%;65~75min甲醇体积82%
→
84%,0.08%磷酸体积8%
→
6%;75~90min甲醇体积84%
→
86%,0.08%磷酸体积6%
→
4%; 90~100min甲醇体积86%
→
88%,0.08%磷酸体积4%
→
2%;100~110min 甲醇体积88%
→
90%,0.08%磷酸体积2%
→
0%时可实现色谱图中各色谱峰的良好分离度。
[0055]
下面将结合实施例对本发明的实施方案进行详细描述,实施例中未注明具体条件者,按照常规条件或制造商建议的条件进行。所用试剂或仪器未注明生产厂商者,均为可以通过市购而获得的常规产品。
[0056]
实施例用到的仪器与试剂如下:
[0057]
实验器材
[0058]
1.仪器如表2
[0059]
表2本发明采用的仪器
[0060][0061][0062]
2.药品与试剂
[0063]
11批次西黄胶囊样品批号如下表3。
[0064]
表3本发明西黄胶囊样品批号
[0065][0066]
对照品:胆酸对照品(批号ly0307);猪去氧胆酸(批号ly0686);去氧胆酸对照品(批号ly0306);胆红素对照品(批号ly0305);麝香酮对照品(批号ly0810);没药酮对照品(批号pcs1410);山达海松酸对照品(批号r19618);苦木素对照品(批号btq509400);11-羰基-β-乳香酸对照品(批号ly0864); 11-羰基-β-乙酰乳香酸对照品(批号ds1195);乙酰11α-甲氧基-β-乳香酸对照品(批号ha015687),以上对照品均购买于中国食品药品检定研究所;甲醇(分析纯);磷酸(分析纯);乙腈(色谱纯)。
[0067]
实施例1一种西黄胶囊的指纹图谱构建方法,包括以下步骤:
[0068]
s1、西黄胶囊供试品溶液的制备:
[0069]
取11批次的西黄胶囊内容物240mg置250ml具塞锥形瓶中,加[甲醇
‑ꢀ
氯仿(1:3)]-磷酸(100:0.2)溶液100ml,超声提取30min,滤液过0.45μm 微孔滤膜,得供试品溶液;
[0070]
s2、标准品溶液的制备:
[0071]
分别精密称定胆酸、猪去氧胆酸、去氧胆酸、胆红素、麝香酮、没药酮,置于容量瓶中,用乙醇定容至刻度,得混合标准品溶液1;精密称定苦木素、 11-羰基-β-乳香酸、11-羰基-β-乙酰乳香酸、乙酰-11α-甲氧基-β-乳香酸、山达海松酸,置于容量瓶中,用甲醇定容至刻度,摇匀,得混合标准品溶液2,制成的混合标准品溶液中胆酸、猪去氧胆酸、去氧胆酸、胆红素、麝香酮、没药酮、苦木素、11-羰基-β-乳香酸、11-羰基-β-乙酰乳香酸、乙酰-11α-甲氧基-β
‑ꢀ
乳香酸、山达海松酸的浓度分别为200μg/ml、200μg/ml、200μg/ml、40μg/ml、 150μg/ml、150μg/ml、100μg/ml、100μg/ml、100μg/ml、100μg/ml、 150μg/ml。
[0072]
s3、分别精密吸取11批西黄胶囊供试品溶液和标准品溶液,注入高效液相色谱仪,记录色谱图;液相色谱条件为:色谱柱:hypersil c18 ods(4.6
×
250 mm
×
5μm);流动相:甲醇(a)-0.08%磷酸(b)-乙腈(c),梯度洗脱;紫外可见吸收检测器多波长切换检测,检测波长:0~15min:254nm;15~45 min:249nm;45~90min:239nm;90~110min:210nm;柱温:27℃;流速0.6ml/min;进样体积:20μl;洗脱程序如下表1。
[0073]
s4、将s3中获得的11批西黄胶囊供试品溶液的色谱图导出,并导入中药色谱指纹图谱相似度评价系统2004a;选择11批次西黄胶囊的色谱图中均存在的色谱峰作为共有峰;
用平均值计算法生成西黄胶囊的对照指纹图谱r,计算各共有峰的相对保留时间和相对峰面积;并根据标准品溶液色谱图的保留时间标注对照指纹图谱中峰的化学成分;
[0074]
s5、将s3中获得的西黄胶囊供试品溶液的色谱图(图3)和标准品溶液色谱图(图1和图2)进行比对,并结合图4-图10,指认主要成分,对比出西黄胶囊中3、4、8、10、11、12、13号色谱峰分别为:没药酮(保留时间18.834min)、山达海松酸(保留时间22.316min)、苦木素(保留时间33.579min)、麝香酮 (保留时间44.632min)、11-羰基-β-乳香酸(保留时间49.086min)、11-羰基-β
‑ꢀ
乙酰乳香酸(保留时间53.643min)、乙酰-11α-甲氧基-β-乳香酸(保留时间 58.967min),得到西黄胶囊的指纹图谱。
[0075]
同时本发明使用自动生成的对照指纹图谱r来生成共有色谱峰模式,分析计算获得11批西黄胶囊共有色谱峰之间具有相对很好的相似性,说明该方法建立的西黄胶囊的指纹图谱能够很好的检测西黄胶囊及11批次西黄胶囊的质量,结果如表4。
[0076]
表4各批次样品与共有色谱峰模式间的相似度
[0077][0078][0079]
实施例2指纹图谱检测方法的方法学研究:
[0080]
s1、精密度研究
[0081]
取实施例1方法制备的标准品溶液,按照实施例1的检测方法分析,平行进样6次,进样量为20μl,通过分析峰面积及保留时间并计算rsd值,结果见表5,结果表明该设备的平行进样精密性良好。
[0082]
表5精密度研究峰面积和保留时间
[0083][0084]
s2、稳定性研究
[0085]
取西黄胶囊内容物1.25g按实施例1方法制备供试品溶液,按照实施例1 的检测方法分析,采用0、2、6、12、18、24h不同时间进样分析,进样量为 20μl,以没药酮、山达海松酸、苦木素、麝香酮、11-羰基-β-乳香酸、11-羰基-β-乙酰乳香酸、乙酰-11α-甲氧基-β-乳香酸为参照峰,通过分析样品hplc指纹图谱的共有峰的峰面积及保留时间并计算rsd值,结果见表6,表明西黄胶囊供试品溶液在24h之内的色谱峰几乎没有变化,稳定性好。
[0086]
表6稳定性研究峰面积和保留时间
[0087][0088]
s3、重复性研究
[0089]
按实施例1中供试品溶液方法制备六批样品溶液,参照实施例1的色谱条件,进样量为20μl,以没药酮、山达海松酸、苦木素、麝香酮、11-羰基-β-乳香酸、11-羰基-β-乙酰乳香酸、乙酰-11α-甲氧基-β-乳香酸为参照峰,通过分析样品hplc指纹图谱的共有峰的峰面积及保留时间并计算rsd值,结果见表4,结果表明样品色谱峰重现性好,该方法的重复性良好。
[0090]
表7重复性研究峰面积和保留时间
[0091][0092]
以上实验结果表明,本发明提供的西黄胶囊的指纹图谱构建方法,具有稳定性好,精密度高,重复性好的特点,能全面客观评价西黄胶囊的质量,为临床疗效提供质量保证。
[0093]
以上实施例仅为本发明的示例性实施例,不用于限制本发明,本发明的保护范围由权利要求书限定。以上内容仅作为对本发明的构思所作的举例和说明,所属本技术领域的技术人员对所描述的具体实施例所做各种的修改或补充或采用类似的方式替代,只要不偏离发明的构思或者超越权利要求书所定义的范围,均应属于本发明的保护范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1