一种夏桑菊颗粒定性和定量检测方法与流程
1.本发明涉及中药质量检测技术领域,具体是一种夏桑菊颗粒定性和定量检测方法。
背景技术:
2.夏桑菊颗粒是由夏枯草、桑叶、野菊花三味药材按500:175:80(w/w/w)的比例配伍组成的一种中药制剂,收录于中华人民共和国药典(2020版)一部,也常作为凉茶饮料。据记载,夏桑菊颗粒具有清肝明目、疏风散热、除湿痹、解疮毒的功效,用于风热感冒、目赤头痛、头晕耳鸣、咽喉肿痛、疔疮肿毒等症状。现代药理研究表明,它还具有抗氧化、抗炎、抗病毒、抗癌、免疫调节活性和心血管保护等作用。
3.近年来,有诸多夏桑菊颗粒质量控制的相关研究。在定性鉴别方面,中国药典除对照药材外,还采用迷迭香酸和蒙花苷作为指标成分鉴别夏枯草和野菊花,但桑叶的鉴别仅采用对照药材,缺乏专属性指标成分。另外,药典中采用3个展开系统分别对夏枯草、桑叶、野菊花进行鉴别,鉴别方法相对复杂。专利号为cn 103018391 b的专利涉及“一种夏桑菊颗粒的质量控制方法”简化了鉴别条件,选择对照药材和迷迭香酸作为对照品在一块薄层板上同时检测三味药材,但该法缺乏桑叶和野菊花的专属性指标成分,且展开剂中使用了三氯甲烷,毒性较大。
4.在含量测定方面,中国药典中仅测定迷迭香酸的含量。然而考虑到中药制剂成分的复杂性,单一成分的定量分析无法全面、准确、有效地控制该制剂的质量。林丽美等人采用《rp-hplc法同时测定夏桑菊颗粒中绿原酸、异迷迭香酸苷、迷迭香酸和蒙花苷》(中成药,2013年11期);孙鹏等人在文献《夏桑菊凉茶中多成分含量测定方法的研究》(食品工业,2016年4期)中采用高效液相色谱法对夏桑菊凉茶中的绿原酸、迷迭香酸、木犀草苷、芦丁、槲皮素和蒙花苷6种活性成分进行了定量分析;桑岚在文献《夏桑菊颗粒不同干燥方式对指标成分含量的影响》(中医学报,2019年34期)中通过测定绿原酸,迷迭香酸和蒙花苷的含量探究不同干燥方式对制剂的质量影响。多成分含量测定虽然可以较好地反映制剂的质量,但外标法需要对照品种类多,对照品消耗量大,检测经济成本较高,限制了其在实际工作中的应用。
5.一测多评法是由王智民等人在2006年率先提出的多指标质量控制方法,该方法是利用中药有效成分内在的函数关系和比例关系,以一种相对易得、廉价的对照品为内参物,实现对多个成分的同时测定。一测多评法具有检测成本低,分析效率高的优点,目前已经应用于丹参、淫羊藿、银杏叶胶囊、咳特灵片等中药材和中药制剂的中国药典质量标准中。但是,尚未见用一测多评法同时测定夏桑菊颗粒中多种成分含量的报道。
6.因此,本发明致力于简化定性鉴别实验,优化鉴别条件,实现同时鉴别夏桑菊颗粒中的三味药材,并识别其专属性指标成分。同时为了节省检测成本,提高定量分析效率,本发明还采用双内参一测多评法同时测定夏桑菊颗粒中多种活性成分,从而建立一种简易且全面的质量评价方法。
技术实现要素:
7.本发明目的在于提供一种夏桑菊颗粒定性和定量检测方法。
8.所述的定性检测方法采用薄层色谱法,在同一薄层板上实现了夏枯草、桑叶和野菊花三味中药的同时鉴别,并识别其专属性指标成分迷迭香酸、东莨菪苷和蒙花酸,简化了鉴别步骤,提高了鉴别效率。
9.所述的定量检测方法采用超高效液相色谱技术,结合双内参一测多评法,以绿原酸和迷迭香酸为内参物,同时测定夏桑菊颗粒中新绿原酸、绿原酸、隐绿原酸、咖啡酸、异迷迭香酸苷、异绿原酸b、异绿原酸a、迷迭香酸、蒙花苷9种成分的含量,节省了检测成本,方法准确可靠,重复性良好,且与外标法测定结果无显著性差异,为市面上质量参差不齐的夏桑菊颗粒产品的质量控制提供参考。
10.为实现上述目的,本发明是通过以下技术方案实现的:一种夏桑菊颗粒定性和定量检测方法,所述得检测方法包括薄层色谱定性鉴别和超高效液相色谱定量测定;所述夏桑菊颗粒薄层色谱定性测定是以迷迭香酸、东莨菪苷、蒙花苷为对照品,将夏枯草、桑叶、野菊花的对照药材溶液、对照品溶液、供试品溶液在同一硅胶薄层板上成条带状点样,经展开和显色后得到薄层色谱图,实现对夏桑菊颗粒的定性鉴别,具体步骤如下:(1)供试品溶液的制备;(2)对照药材溶液的制备;(3)对照品溶液的制备;(4) 薄层色谱鉴别:吸取供试品溶液,对照药材溶液和对照品溶液,分别点于同一硅胶gf
254
高效薄层板上成条带状,以体积比为3:15:2:1的甲苯-乙酸乙酯-甲酸-水为展开剂展开,取出晾干后,置紫外光灯366 nm下检视;之后100-105℃加热薄层硅胶板3-5分钟,先喷以浓度为1%的2-氨基乙基联苯基硼酸酯甲醇溶液,再喷以5%的聚乙二醇乙醇溶液,晾干后,置于紫外光灯366 nm下检视。
11.进一步地,上述步骤(1)中所述的供试品溶液的制备方法为:取夏桑菊颗粒10 g,研细,加无水乙醇30ml,超声处理30分钟,滤过,滤液旋干,残渣溶解于2ml无水乙醇,作为供试品溶液;进一步地,上述步骤(2)中所述的对照药材溶液的制备方法为:取夏枯草对照药材0.5g、桑叶对照药材1.0g、野菊花对照药材1.0g,分别加15ml无水乙醇超声处理30分钟,滤过,滤液旋干,残渣溶解于2ml无水乙醇,作为对照药材溶液;进一步地,上述步骤(3)中所述的对照品溶液的制备方法为:取迷迭香酸、东莨菪苷、蒙花苷对照品,加甲醇制成每1ml含以上对照品各0.3-0.5 mg的混合溶液,作为对照品溶液。
12.所述夏桑菊颗粒超高效液相色谱含量测定是以绿原酸内参物,计算新绿原酸、隐绿原酸、咖啡酸的相对保留时间和相对校正因子;以迷迭香酸为内参物,计算异迷迭香酸苷、异绿原酸b、异绿原酸a、蒙花苷的相对保留时间和相对校正因子,采用双内参一测多评法同时测定夏桑菊颗粒中新绿原酸、绿原酸、隐绿原酸、咖啡酸、异迷迭香酸苷、异绿原酸b、异绿原酸a、迷迭香酸、蒙花苷9种成分的含量,从而实现对夏桑菊颗粒的定量分析。
13.具体内容如下:(a) 对照品溶液的制备;(b) 供试品溶液的制备;(c) 色谱条件:采用c
18
反相色谱柱,其规格为2.1 mm
×
100 mm,粒径小于2μm;以乙腈(a)和1%乙酸水溶液(b)作为流动相,按如下洗脱程序进行梯度洗脱:0-4 min,8%-10% a;4-6 min,10%-19.5% a;6-13 min,19.5% a;13-18 min,19.5%-35% a;18-20 min,35%-90% a;流速为0.4 ml/min,柱温为30 ℃,检测波长为325 nm,进样量为2 μl;(d) 相对校正因子和相对保留时间的计算:以绿原酸内参物,计算新绿原酸、隐绿原酸、咖啡酸的相对保留时间和相对校正因子;以迷迭香酸为内参物,计算异迷迭香酸苷、异绿原酸b、异绿原酸a、蒙花苷的相对保留时间和相对校正因子;(e) 测定:测定夏桑菊颗粒中绿原酸和迷迭香酸的含量,依据相对校正因子计算新绿原酸、隐绿原酸、咖啡酸、异迷迭香酸苷、异绿原酸b、异绿原酸a、蒙花苷7种成分的含量。
14.进一步地,上述步骤(a)中所述的对照品溶液的制备方法为:分别取新绿原酸、绿原酸、隐绿原酸、咖啡酸、异迷迭香酸苷、异绿原酸b、异绿原酸a、迷迭香酸、蒙花苷适量,精密称定,加甲醇溶解,得到对照品溶液;再进一步地,所述的对照品溶液中新绿原酸、绿原酸、隐绿原酸、咖啡酸、异迷迭香酸苷、异绿原酸b、异绿原酸a、迷迭香酸、蒙花苷的质量浓度分别为0.192 mg/ml、0.192 mg/ml、0.185 mg/ml、0.131 mg/ml、0.160 mg/ml、0.162 mg/ml、0.135 mg/ml、0.262 mg/ml和0.198 mg/ml。
15.进一步地,上述步骤(b)中所述的供试品溶液的制备方法为:取夏桑菊颗粒研细粉末1.25 g,加50%乙醇溶液25ml,超声处理30分钟,冷却至室温,补足失重,摇匀,3500 rpm离心10分钟,取上清液,用0.22μm滤膜过滤,取续滤液作为供试品溶液。
16.进一步地,上述步骤(c)中所述的c
18
反相色谱柱为waters acquity uplc hss t3 (100 mm
×
2.1 mm, 1.8 μm)、waters acquity uplc cortecs t3 (100 mm
×
2.1 mm, 1.6 μm)、waters acquity uplc beh c18 (100 mm
×
2.1 mm, 1.7 μm)或thermo scientific hypersil gold (100 mm
×
2.1 mm, 1.9 μm)中的一种;优选为waters acquity uplc hss t3 (100 mm
×
2.1 mm,1.8 μm)。
17.进一步地,上述步骤(d)中所述的绿原酸对于新绿原酸、隐绿原酸、咖啡酸的相对保留时间分别为0.561-0.635、1.120-1.343、1.301-1.355;优选为0.561、1.153、1.309;所述的迷迭香酸对于异迷迭香酸苷、异绿原酸b、异绿原酸a、蒙花苷的相对保留时间分别为0.779-0.866、0.815-0.888、0.855-0.906、1.522-1.699;优选为0.787、0.815、0.855、1.547。
18.进一步地,所述绿原酸对于新绿原酸、隐绿原酸、咖啡酸的相对校正因子分别为0.788-0.884、0.894-0.927、0.462-0.517,优选为0.804、0.904、0.499;所述的迷迭香酸对于异迷迭香酸苷、异绿原酸b、异绿原酸a、蒙花苷的相对校正因子分别为1.269-1.403、1.276-1.444、0.659-0.747、1.075-1.272,优选为1.352、1.321、0.671、1.152。
19.与现有技术相比本发明的有益效果在于:
(1)在定性方面,本发明优化了展开条件,识别了夏枯草、桑叶、野菊花的专属性指标成分迷迭香酸、东莨菪苷和蒙花苷,在一块薄层板上实现了夏桑菊颗粒中三味中药材的同时鉴别,简化了鉴别步骤,从而提高了鉴别效率,促进了夏桑菊颗粒的整体质量控制。
20.(2)在定量方面,为节省检测成本和提高分析效率,本发明采用了超高效液相色谱法结合双内参一测多评法同时测定夏桑菊颗粒中新绿原酸、绿原酸、隐绿原酸、咖啡酸、异迷迭香酸苷、异绿原酸b、异绿原酸a、迷迭香酸、蒙花苷9种活性成分的含量,方法准确可靠,重复性良好,且与外标法测定结果无显著性差异,为夏桑菊颗粒的质量评价提供了一种全面且综合的测定方法。
附图说明
21.为了更清楚地说明本发明实施例的技术方案,下面将对实施例中所需要使用的附图作简单地介绍,应当理解,以下附图仅示出了本发明的某些实施例,因此不应被看作是对范围的限定,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其他相关的附图。
22.图1为混合对照品(ms1和ms2)和夏桑菊颗粒样品(s16)采用不同展开系统的薄层色谱图;a:甲苯-乙酸乙酯-甲酸-乙酸-水(1:15:1:1:2, v/v/v/v/v);b:甲苯-乙酸乙酯-甲酸-水 (3:15:2:1, v/v/v/v)。
23.其中1:芦丁;2:东莨菪苷;3:蒙花苷;4:绿原酸;5:金丝桃苷;6:木犀草苷;7:异槲皮苷;8:紫云英苷;9:异绿原酸c;10:异绿原酸b;11:异绿原酸a;12:迷迭香酸;13:东莨菪素;14:咖啡酸。
24.图2为混合对照品(ms1和ms2)和夏桑菊颗粒样品(s16)采用不同显色方式的薄层色谱图;a:直接在紫外光灯254 nm下检视;b:直接在紫外光灯366 nm下检视;c:喷以5%三氯化铁乙醇溶液,105℃加热3分钟后在白光下检视;d:喷以1%三氯化铝乙醇溶液,105℃加热5分钟后在紫外光灯366 nm下检视;e:100-105℃加热薄层硅胶板3-5分钟,喷以浓度为1%的2-氨基乙基联苯基硼酸酯甲醇溶液,再喷以5%的聚乙二醇乙醇溶液,晾干后,在紫外灯366 nm下检视。
25.其中1:芦丁;2:东莨菪苷;3:蒙花苷;4:绿原酸;5:金丝桃苷;6:木犀草苷;7:异槲皮苷;8:紫云英苷;9:异绿原酸c;10:异绿原酸b;11:异绿原酸a;12:迷迭香酸;13:东莨菪素;14:咖啡酸。
26.图3为混合对照品(ms1和ms2)、对照药材(rd1-rd3)和夏桑菊颗粒样品(s1-s16)的薄层色谱图。
27.其中,rd1:夏枯草对照药材;rd2:桑叶对照药材;rd3:野菊花对照药材;1:芦丁;2:东莨菪苷;3:蒙花苷;4:绿原酸;5:金丝桃苷;6:木犀草苷;7:异槲皮苷;8:紫云英苷;9:异绿原酸c;10:异绿原酸b;11:异绿原酸a;12:迷迭香酸;13:东莨菪素;14:咖啡酸。
28.图4为对照品(r1-r3)、单味药材(d1-d3)和夏桑菊颗粒样品(s9)的薄层色谱图,其中,d1:桑叶药材;d2:野菊花药材;d3:夏枯草药材;1:芦丁;2:蒙花苷;3:绿原酸;4:异槲皮苷;5:紫云英苷;6:木犀草苷;7:异绿原酸a;8:异绿原酸b;9:异绿原酸c;10:迷
迭香酸。
29.图5为混合对照品溶液(a)和夏桑菊颗粒供试品溶液(b)的超高效液相色谱图,其中,1:新绿原酸;2:绿原酸;3:隐绿原酸;4:咖啡酸;5:异迷迭香酸苷;6:异绿原酸b;7:异绿原酸a;8:迷迭香酸;9:蒙花苷。
30.图6为夏桑菊颗粒供试品溶液的高效液相色谱图,其中,1.新绿原酸;2.绿原酸;3.隐绿原酸;4.咖啡酸;5.异迷迭香酸苷;6.异绿原酸b;7.异绿原酸a;8.迷迭香酸;9.蒙花苷。
具体实施方式
31.为使本发明实施例的目的、技术方案和优点更加清楚,下面将对本发明实施例中的技术方案进行清楚、完整地描述。实施例中未注明具体条件者,按照常规条件或制造商建议的条件进行。所用试剂或仪器未注明生产厂商者,均为可以通过市售购买获得的常规产品。
32.实施例1:夏桑菊颗粒的薄层色谱鉴别1. 仪器与试药1.1 仪器薄层色谱自动点样系统automatic tlc sampler4 (camag, switzerland)、薄层色谱自动展开系统adc2 (camag , switzerland)、薄层色谱数码成像仪tlc visualizer 2 (camag, switzerland)、薄层色谱加热板tlc plate heater
ꢀⅲꢀ
(camag, switzerland);梅特勒托利多xs105 型电子分析天平 (十万分之一,梅特勒-托利多国际贸易(上海)有限公司),必能信8510 超声波清洗机(美国branson超声波科技有限公司)。
33.1.2 材料和试剂高效硅胶薄层板gf
254 (20
×
10cm)购自于德国默克股份有限公司。实验试剂均为分析纯。2-氨基乙基二苯硼酸盐和聚乙二醇400购自德国卡尔罗斯公司(karlsruhe, germany),醋酸(glacial, 100%)购自德国默克公司(darmstadt, germany),甲苯(gr, acs 99.5%)购自美国国际实验室(south san francisco, ca, usa),氯化铁、氯化铝晶体购于中国天津大茂化学试剂厂,其他试剂如甲酸、乙酸乙酯、甲醇和乙醇购于广东西陇科学股份有限公司。水为millipore milli q-plus超纯水系统 (millipore, billerica, ma, usa)制备所得。
34.1.3 对照品中药对照品均为hplc级,纯度均在98%以上。绿原酸、异绿原酸a、异绿原酸b、异绿原酸c、咖啡酸、迷迭香酸、蒙花苷、芦丁、金丝桃苷、东莨菪苷、东莨菪素购于中国成都曼思特生物科技有限公司。紫云英苷、木犀草苷购自成都普菲德生物技术有限公司,异槲皮苷购自成都普瑞法生物技术有限公司。夏枯草对照药材(批号120993-202007)、桑叶对照药材(批号121123-201806)和野菊花对照药材(批号120995-201707)均购自中国食品药品检定研究院(北京)。
35.1.4 样品不同厂家夏桑菊颗粒采购于各地药房,详细信息见表1。
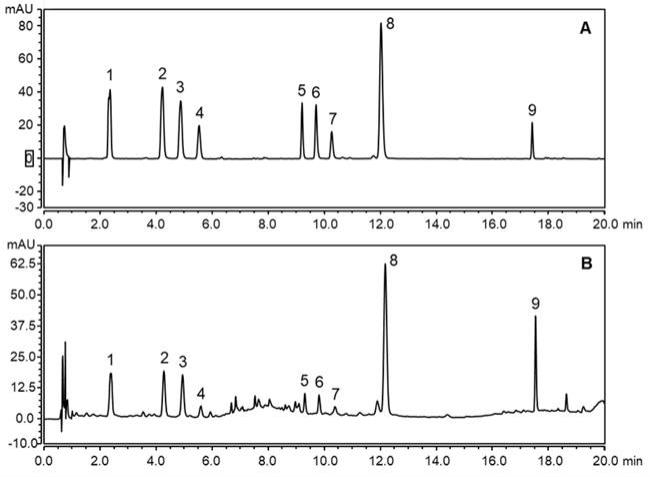
36.表1夏桑菊颗粒样品信息
样品编号生产厂家批号s1其他公司a200501s2其他公司b200704s3其他公司c200615s4其他公司d210403s5其他公司e201219s6其他公司e200804s7其他公司e200904s8广州白云山星群(药业)股份有限公司sa10053s9广州白云山星群(药业)股份有限公司ra10187s10广州白云山星群(药业)股份有限公司rc90138s11其他公司f210502s12其他公司g200313s13其他公司g210206s14其他公司h20201102s15其他公司i20200702s16其他公司j1906182. 方法与结果2.1 供试品溶液的制备取夏桑菊颗粒10 g,研细,加无水乙醇30ml,超声处理30分钟,滤过,滤液旋干,残渣溶解于2ml无水乙醇,作为供试品溶液;2.2对照药材溶液的制备取夏枯草对照药材0.5 g、桑叶对照药材1.0 g、野菊花对照药材1.0 g,分别加15ml无水乙醇超声处理30分钟,滤过,滤液旋干,残渣溶解于2ml无水乙醇,作为对照药材溶液;2.3 对照品溶液的制备取芦丁、东莨菪苷、蒙花苷、绿原酸、金丝桃苷、异槲皮苷、木犀草苷、紫云英苷、异绿原酸a、异绿原酸b、异绿原酸c、迷迭香酸、东莨菪素和咖啡酸对照品,加甲醇制成每1ml含以上对照品各0.3-0.5 mg的混合溶液,作为对照品溶液;2.4 薄层色谱鉴别吸取供试品溶液,对照药材溶液和对照品溶液2-8μl,分别点于同一硅胶gf
254
高效薄层板上成条带状,以体积比为3:15:2:1的甲苯-乙酸乙酯-甲酸-水为展开剂,展开,取出晾干后,置紫外光灯366nm下检视,供试品色谱中,在与对照药材色谱相应的位置上,显相同颜色的荧光主条带;在与迷迭香酸、东莨菪苷对照品色谱相应的位置上,显相同的蓝色荧光条带;之后100-105℃加热薄层硅胶板3-5分钟,先喷以浓度为1%的2-氨基乙基联苯基硼酸酯甲醇溶液,再喷以5%的聚乙二醇乙醇溶液(np/peg),晾干后,置于紫外光灯366nm下检视,供试品色谱中,在与对照药材色谱相应的位置上,显相同颜色的荧光主条带;在与蒙花苷对照品色谱相应的位置上,显相同的黄色荧光条带;在与迷迭香酸对照品色谱相应的位置上,显相同的青绿色荧光条带。
37.2.5 对照品的选择为选择合适的专属性指标成分,我们选择了14种对照品进行考察。根据文献报道以及与对照药材的比对,夏枯草中含有芦丁、绿原酸、金丝桃苷、异槲皮苷、木犀草苷、迷迭香酸、咖啡酸,桑叶中含有芦丁、东莨菪苷、绿原酸、异槲皮苷、紫云英苷、东莨菪素,野菊花中含有芦丁、蒙花苷、绿原酸、金丝桃苷、异槲皮苷、木犀草苷、紫云英苷、异绿原酸a、异绿原酸b、异绿原酸c、咖啡酸。综合考虑对照品的专属性、薄层色谱的分离情况(图1-3)、各成分在夏桑菊颗粒中的含量以及实际应用中的可行性和对照品的成本,最终选择迷迭香酸、东莨菪苷和蒙花苷分别为夏枯草、桑叶和野菊花的专属性指标成分。
38.2.6 展开系统的考察我们分别考察了甲苯-乙酸乙酯-甲酸-乙酸-水(1:15:1:1:2, v/v/v/v/v)和甲苯-乙酸乙酯-甲酸-水(3:15:2:1, v/v/v/v),两种展开剂分离情况类似,但后者为四相体系,比前者少用一种试剂,故选择后者作为展开系统。在此条件下,东莨菪苷和蒙花苷;异槲皮苷和木犀草苷;异绿原酸b和异绿原酸c;东莨菪素和咖啡酸未能分离,为便于比对,14种对照品分为两组配制混合对照品溶液(图1)。
39.2.7 显色方式的考察我们还考察了直接在紫外光灯254nm和366nm下检视;喷以5%三氯化铁乙醇溶液,105℃加热3分钟后在白光下检视;喷以1%三氯化铝乙醇溶液,105℃加热5分钟后在紫外光灯366nm下检视; 100-105℃加热薄层硅胶板3-5分钟,喷以np/peg显色剂,在紫外灯366nm下检视几种检视方式,结果见图2。在254nm下,东莨菪素和东莨菪苷显蓝色荧光,其它对照品均为暗斑,无法根据颜色区分不同结构类型的化合物。在366nm下,东莨菪苷、绿原酸、异绿原酸a、异绿原酸b、异绿原酸c、迷迭香酸、东莨菪素、咖啡酸显蓝色荧光,其它对照品不显色。喷显色剂5%三氯化铁乙醇溶液后,除东莨菪素和东莨菪苷外,其它对照品均显棕褐色,但灵敏度较低,样品中只有蒙花苷、迷迭香酸和咖啡酸的条带较清晰,其它条带难以观察到。喷显色剂1%三氯化铝乙醇溶液和np/peg后均能使所有对照品显示不同颜色的荧光条带,但后者比前者颜色更丰富,且灵敏度更高,故最终选择了np/peg显色剂。由于东莨菪苷和蒙花苷未能分开,显色前在366nm下蒙花苷不显色而东莨菪苷显蓝色荧光,显色后蒙花苷显黄色荧光而东莨菪苷条带在样品中很微弱几乎被掩盖,可据此辨认这两个成分。因此,最终选择先在紫外光灯366nm下直接检视,辨认迷迭香酸和东莨菪苷,再喷以np/peg显色剂,在紫外光灯366 nm下检视,辨认蒙花苷,同时确认迷迭香酸。
40.2.8 样品测定采用优化后的薄层色谱鉴别方法,对收集到的16批夏桑菊颗粒进行薄层色谱鉴别分析(图3)。结果显示,所有样品中均能观察到清晰的迷迭香酸和东莨菪苷条带,但有一批样品中观察不到蒙花苷条带(s14)。从薄层色谱图来看,市面上流通的夏桑菊颗粒质量差异明显。
41.对比例1按照中国专利申请cn103018391b中公开的“一种夏桑菊颗粒的质量控制方法”中提供的薄层色谱鉴别方法进行检测:展开剂:氯仿:乙酸乙酯:丙酮:甲酸(25:15:10:2)显色剂:1%三氯化铝乙醇溶液,105℃加热5分钟。
42.检测结果见附图4。
43.根据图4的检测结果可以看出:采用现有的展开剂进行有效成分检测时,迷迭香酸的rf值为0.41,位置合适,分离度佳。但其它对照品的rf值均小于0.2,且蒙花苷、绿原酸和异槲皮苷无法分离,异绿原酸a、b、c也无法分离。与单味药材比对时,在夏桑菊颗粒的薄层图谱中虽可检测出夏枯草药材中主要条带,但与桑叶和野菊花药材相比无明显相同条带。因此,该方法可清晰地鉴别夏枯草,但对桑叶和野菊花的鉴别效果不佳。而在本技术的方法中,所有对照品的rf值均在0.1-0.9之间,分离效果较好,且指认了迷迭香酸、东莨菪苷和蒙花苷分别为夏枯草、桑叶和野菊花的专属性指标成分,可同时鉴别夏桑菊颗粒的三味组成药材。
44.实施例2:夏桑菊颗粒的双内参一测多评含量测定1. 仪器与试药1.1 仪器赛默飞 ultimate
™ꢀ
3000型超高效液相色谱系统(thermo fisher scientific inc., waltham, ma, usa);沃特世 acquity uplc i-class 超高效液相色谱仪(waters, milford, ma, usa);梅特勒托利多xs105 型电子分析天平 (十万分之一,梅特勒-托利多国际贸易(上海)有限公司);必能信8510 超声波清洗机(美国branson超声波科技有限公司);赛默飞heraeus multifuge x3r型离心机(thermo fisher scientific inc.,waltham,ma,usa)。
45.1.2 材料和试剂色谱柱:waters acquity uplc hss t3 (100 mm
×
2.1 mm, 1.8 μm)、waters acquity uplc cortecs t3 (100 mm
×
2.1 mm, 1.6 μm)、waters acquity uplc beh c18 (100 mm
×
2.1 mm, 1.7 μm)和thermo scientific hypersil gold (100 mm
×
2.1 mm, 1.9 μm)。hplc级乙腈和甲醇购自aci labscan有限公司(bangkok, thailand)。醋酸(glacial, 100%)购自德国默克公司(darmstadt, germany),分析纯甲醇和乙醇购于中国天津大茂化学试剂厂。水为millipore milli q-plus超纯水系统 (millipore, billerica, ma, usa)制备所得。
46.1.3 对照品和样品中药对照品均为hplc级,纯度均在98%以上。隐绿原酸、绿原酸、新绿原酸、异迷迭香酸苷、咖啡酸、异绿原酸a、异绿原酸b、迷迭香酸和蒙花苷购于中国成都曼思特生物科技有限公司。不同厂家夏桑菊颗粒采购于各地药房,详细信息见表1。
47.2. 方法与结果2.1 色谱条件色谱柱:waters acquity uplc hss t3 column (2.1 mm
ꢀ×ꢀ
100 mm, 1.8 μm);以乙腈(a)和1%乙酸水溶液(b)作为流动相,按如下洗脱程序进行梯度洗脱:0-4 min,8%-10% a;4-6 min,10%-19.5% a;6-13 min,19.5% a;13-18 min,19.5%-35% a;18-20 min,35%-90% a;流速0.4ml/min;柱温30℃;检测波长为325nm;进样量2μl。
48.2.2 对照品溶液制备分别取新绿原酸、绿原酸、隐绿原酸、咖啡酸、异迷迭香酸苷、异绿原酸b、异绿原酸a、迷迭香酸、蒙花苷适量,精密称定,加甲醇溶解,制备成含新绿原酸、绿原酸、隐绿原酸、咖
啡酸、异迷迭香酸苷、异绿原酸b、异绿原酸a、迷迭香酸、蒙花苷的质量浓度分别为0.192 mg/ml、0.192 mg/ml、0.185 mg/ml、0.131 mg/ml、0.160 mg/ml、0.162 mg/ml、0.135 mg/ml、0.262 mg/ml和0.198 mg/ml的混合对照品溶液。
49.2.3 供试品溶液的制备取夏桑菊颗粒研细粉末1.25 g,精密称定,加入50%乙醇25 ml,超声处理30分钟,冷却至室温,补足失重,摇匀,3500 rpm离心10分钟,取上清液,用0.22μm滤膜过滤,取续滤液作为供试品溶液。
50.2.4 方法学考察2.4.1 线性范围、检测限和定量限精密吸取“2.2”项混合对照品溶液1.5 ml、1.2 ml、1.0 ml、0.8 ml、0.6 ml、0.4 ml、0.2 ml、0.1 ml分别置于2 ml容量瓶中,用甲醇稀释至刻度,摇匀,按“2.1”项色谱条件进样测定,分别以各对照品质量浓度为横坐标(x),峰面积为纵坐标(y),绘制标准曲线,得到回归方程、线性范围和相关系数。将混合对照品溶液逐级稀释,以信噪比为3:1时为检测限,以信噪比为10:1时为定量限。新绿原酸、绿原酸、隐绿原酸、咖啡酸、异迷迭香酸苷、异绿原酸b、异绿原酸a、迷迭香酸、蒙花苷的线性回归方程、相关系数、线性范围、检测限和定量限结果见表2。表2 标准曲线、检测限和定量限2.4.2 精密度精密吸取线性范围内的三个浓度的混合对照品溶液,在一天内连续进样6次,以及在3天每天进样2次,记录峰面积,用峰面积的rsd值来评价日内、日间精密度,结果表明仪器精密度良好(表3)。
51.表3 日间和日内精密度结果
2.4.3 重复性精密称取夏桑菊颗粒(s8)研细粉末1.00 g、1.25 g、1.50 g,按“2.3”项下方法制备供试品溶液,每个浓度水平平行3份,按“2.1”项色谱条件进样测定,记录峰面积,计算各峰峰面积的rsd值,结果表明方法的重复性良好(表4)。
52.2.4.4 稳定性精密称取夏桑菊颗粒(s8)研细粉末1.25 g,按“2.3”项下方法制备供试品溶液,按“2.1”项色谱条件,分别在0、2、4、8、12、24、48 h进样,记录峰面积,计算各峰峰面积的rsd值,结果表明供试品溶液在48h内稳定(表4)。
53.表4 重复性和稳定性结果
注:+:低于定量限。
54.2.4.5 加样回收率试验精密称量夏桑菊颗粒(s8)研细粉末0.65 g,分别按照各成分已知含量的50%、100%、150%加入各对照品适量,按“2.3”项下方法制备供试品溶液,每个浓度水平平行3份,按“2.1”项色谱条件进样测定,计算加样回收率,结果见表5,各成分的加样回收率在95.03%-105.71%内,表明方法的准确性良好。
55.表5 加样回收率结果
2.5 双内参一测多评法参考一测多评法指南,应选择中药制剂中相对易得、含量较高、药理作用相对明确的成分作为内参物。根据实验经验以及文献检索,采用一测多评法进行多组分含量定量分析时,选择的内参物与待测成分的结构越接近,与待测成分的保留时间越接近,计算结果更为准确,也更具有参考意义。考虑到夏桑菊颗粒中9种待测成分结构差异较大,只采用一个内参物无法准确评估各成分,故本实验采用两个内参物进行多成分含量测定,以保证一测多评结果的准确性。迷迭香酸、绿原酸作为夏桑菊颗粒中含量较高的酚酸类成分,稳定性较
好,对照品价格便宜易得,因此本实验选择迷迭香酸和绿原酸作为内参物,同时测定夏桑菊颗粒中9种成分的含量。
56.2.5.1 相对校正因子的确定将“2.4.1”项下不同浓度系列混合对照品溶液按“2.1”项色谱条件进样,记录峰面积。以绿原酸和迷迭香酸为内参物,按公式(1)分别计算各化合物的相对校正因子。结果绿原酸对于新绿原酸、隐绿原酸、咖啡酸的相对校正因子分别为0.804、0.904、0.499;迷迭香酸对于异迷迭香酸苷、异绿原酸b、异绿原酸a以及蒙花苷的相对校正因子分别为1.352、1.321、0.671和1.152。
57.式中:f
si
为内参物(s)对于某待测成分(i)的相对校正因子;as为内参物对照品s峰面积;ai为某待测成分对照品i峰面积;cs为内参物对照品s浓度;ci为某待测成分对照品i浓度。
58.2.5.2 相对校正因子的耐用性考察2.5.2.1 不同仪器和色谱柱对相对校正因子的影响本实验分别考察了waters acquity uplc hss t3 (100 mm
×
2.1 mm, 1.8 μm)、waters acquity uplc cortecs t3 (100 mm
×
2.1 mm, 1.6 μm)、waters acquity uplc beh c18 (100 mm
×
2.1 mm, 1.7 μm)和thermo scientific hypersil gold (100 mm
×
2.1 mm, 1.9 μm) 4根uplc色谱柱以及thermo u3000型和waters i-class型超高效液相色谱仪对各成分的相对校正因子的影响,以绿原酸和迷迭香酸为内参物,分别计算各待测组分的相对校正因子,结果显示各待测成分的相对校正因子的rsd值均小于5%(表6),不同仪器和不同色谱柱对相对校正因子的影响不大。
59.表6 不同仪器和色谱柱对相对校正因子的影响注:c1: 新绿原酸; c2: 绿原酸; c3:隐绿原酸; c4: 咖啡酸; c5: 异迷迭香酸苷; c6: 异绿原酸b; c7:异绿原酸a; c8:迷迭香酸; c9:蒙花苷。
60.2.5.2.2 不同流速、柱温及检测波长对相对校正因子的影响本实验考察了不同柱温(25、30、35℃)、不同流速(0.35、0.40、0.45 ml/min)以及
不同检测波长(320、325、330 nm)对相对校正因子的影响,结果显示各待测成分的相对校正因子rsd值均小于4%,结果表明在不同流速、不同柱温及不同检测波长下测得的各组分相对校正因子无显著性差异(表7)。
61.表7 不同检测波长、流速及柱温对相对校正因子的影响注:c1: 新绿原酸; c2: 绿原酸; c3:隐绿原酸; c4: 咖啡酸; c5: 异迷迭香酸苷; c6: 异绿原酸b; c7:异绿原酸a; c8:迷迭香酸; c9:蒙花苷。
62.2.5.3 色谱峰定位一测多评法应用于多指标成分同时定量可行的前提是对各待测成分色谱峰的准确定位。目前文献中较多采用的方法为相对保留时间定位法或保留时间差值定位法。本实验在thermo u3000型和waters i-class型超高效液相色谱仪上分别用waters acquity uplc hss t3 (100 mm
×
2.1 mm, 1.8 μm)、waters acquity uplc cortecs t3 (100 mm
×
2.1 mm, 1.6 μm)、waters acquity uplc beh c18 (100 mm
×
2.1 mm, 1.7 μm)和thermo scientific hypersil gold (100 mm
×
2.1 mm, 1.9 μm) 4根不同uplc色谱柱考察了各待测组分色谱峰的相对保留时间的重现性;实验结果表明,7个待测成分的相对保留时间的rsd值小于5%,结果见表8。
63.表8 不同液相仪器和色谱柱对色谱峰定位的影响
注:c1: 新绿原酸; c2: 绿原酸; c3:隐绿原酸; c4: 咖啡酸; c5: 异迷迭香酸苷; c6: 异绿原酸b; c7:异绿原酸a; c8:迷迭香酸; c9:蒙花苷。
64.2.6 样品含量测定将收集的16批夏桑菊颗粒按“2.3”项方法制备供试品溶液,每批样品平行3份,按“2.1”项下色谱条件进样,记录各待测成分保留时间及峰面积,分别用外标法(esm)和所建立的双内参一测多评法(qams)计算各成分的含量,并将两种方法的测定结果进行比较,用相对标准偏差rsd值评价两种方法含量测定结果的差异,结果两种方法rsd值小于5%,结果详见表9-1、9-2,混合对照品溶液和代表性样品(s8)的超高效液相色谱图见图5。结果表明双内参一测多评法应用于同时测定夏桑菊颗粒中9种成分具有可行性,方法准确可靠,且能节省检测成本。
65.表9-1 外标法和双内标一测多评法测定夏桑菊颗粒中4种成分含量结果(mg/g,n=3)
注:esm:外标法;qams:一测多评法;+:低于定量限;-:低于检测限表9-2 外标法和双内标一测多评法测定夏桑菊颗粒中5种成分含量结果(mg/g,n=3)注:esm:外标法;qams:一测多评法;+:低于定量限;-:低于检测限对比例2按照中国药典2020版“夏桑菊颗粒”标准中【指纹图谱】项下提供的高效液相色谱
法进行检测,与实施例2的区别在于:2.1 色谱条件色谱柱:agilent zorbax sb-c18 (4.6mm
ꢀ×ꢀ
150mm, 5 μm);以乙腈(a)和1.0%醋酸水溶液(b)作为流动相,按如下洗脱程序进行梯度洗脱:0-50 min,8%-33% a;50-51 min,33%-8% a;51-60 min,8% a;流速:0.9 ml/min;柱温:35℃;检测波长:320nm;进样量10 μl。
66.检测结果见附图6。
67.根据图6可以看出:在此条件下,绿原酸(2)和隐绿原酸(3)未能完全分开,异绿原酸b(6)和异绿原酸a(7)的峰形不好。且采用高效液相色谱柱,分析时间较长,需要约40分钟。而在本技术的方法中,采用超高效液相色谱柱,仅需不到20分钟,且各对照品分离良好。
68.综上,在定性方面,本发明优化了展开条件,识别了夏枯草、桑叶、野菊花的专属性指标成分迷迭香酸、东莨菪苷和蒙花苷,在一块薄层板上实现了夏桑菊颗粒中三味中药材的同时鉴别,简化了鉴别步骤,从而提高了鉴别效率,促进了夏桑菊颗粒的整体质量控制;在定量方面,为节省检测成本和提高分析效率,本发明采用了超高效液相色谱法结合双内参一测多评法同时测定夏桑菊颗粒中新绿原酸、绿原酸、隐绿原酸、咖啡酸、异迷迭香酸苷、异绿原酸b、异绿原酸a、迷迭香酸、蒙花苷9种活性成分的含量,方法准确可靠,重复性良好,且与外标法测定结果无显著性差异,为夏桑菊颗粒的质量评价提供了一种全面且综合的测定方法。
69.以上仅为本发明的优选实施例而已,并不用于限制本发明,对于本领域的技术人员来说,本发明可以有各种更改和变化。凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1