苍耳子与炒苍耳子中药配方颗粒的指纹图谱构建、鉴别与检测方法与流程
1.本发明涉及中药制剂检测领域,特别是涉及一种苍耳子与炒苍耳子中药配方颗粒的指纹图谱构建、鉴别与检测方法。
背景技术:
2.苍耳子为菊科植物苍耳xanthium sibiricum patr.的干燥成熟带总苞的果实,具有散风寒、通鼻窍、祛风湿的功效,是中医临床上治疗鼻炎的首选用药之一。苍耳子主要含有酚酸类、水溶性苷类等化合物,其中酚酸类为具有抗炎抗氧化药理作用的活性成分,而羧基苍术苷和苍术苷为毒性成分。苍耳子有一定的毒性,使用不当可能会导致人体出现恶心呕吐、腹痛腹泻等不良反应。现代研究表明苍耳子炮制后能降低毒性,其中的羧基苍术苷可转化为毒性更低的苍术苷。因此为减少不良反应发生,临床上应该合理使用苍耳子的生品和炮制品。虽然苍耳子饮片和炒苍耳子饮片可以通过外观性状进行鉴别,但是,饮片经过水提、浓缩干燥制成中药配方颗粒后,便失去了药材或饮片原有的外观性状特征,此时采用肉眼或常规技术手段难以对两者进行区分,因此不利于市场上苍耳子和炒苍耳子中药配方颗粒的质量控制。
3.中药化学指纹图谱是中药整体化学成分的表征,可以较为全面地反映中药的化学组分。有传统技术建立了苍耳子药材的uplc指纹图谱,仅能表征苍耳子药材中醇溶性的物质成分,虽然能够应用于苍耳子药材的质量评价与控制,但苍耳子中药配方颗粒在对药材或饮片进行水提、浓缩、干燥、制粒等方法精制后主要包含的是水溶性成分,并且苍耳子在炮制后存在化学成分上的变化,因此该指纹图谱无法准确对苍耳子中药配方颗粒与炒苍耳子中药配方颗粒进行鉴别。
技术实现要素:
4.基于此,本发明提供了一种苍耳子中药配方颗粒或炒苍耳子中药配方颗粒的指纹图谱构建方法,其能够同时表征苍耳子或炒苍耳子中酚酸类成分与水溶性苷类成分,耗时短、操作简单、特征性强、专属性并且重现性好。
5.本发明通过如下技术方案实现。
6.一种苍耳子中药配方颗粒或炒苍耳子中药配方颗粒的指纹图谱构建方法,包括如下步骤:
7.取原儿茶酸、新绿原酸、绿原酸、隐绿原酸、咖啡酸、1,3-o-二咖啡酰奎宁酸、3,5-o-二咖啡酰奎宁酸与4,5-o-二咖啡酰奎宁酸对照品,制备第一混合对照品溶液;取羧基苍术苷三钾盐与苍术苷二钾盐对照品,制备第二混合对照品溶液;
8.取苍耳子中药配方颗粒或炒苍耳子中药配方颗粒,制备苍耳子中药配方颗粒供试品溶液或炒苍耳子中药配方颗粒供试品溶液;
9.分别将所述苍耳子中药配方颗粒供试品溶液或所述炒苍耳子中药配方颗粒供试
品溶液、所述第一混合对照品溶液与所述第二混合对照品溶液进行超高效液相色谱分析,构建所述苍耳子中药配方颗粒的指纹图谱或所述炒苍耳子中药配方颗粒的指纹图谱;
10.其中,超高效液相色谱分析的条件包括:流动相a为乙腈,流动相b为磷酸二氢钠水溶液;采用梯度洗脱;所述梯度洗脱的程序包括:0min~10min,流动相a的体积百分数从2.5%变化至10%;10min~20min,流动相a的体积百分数从10%变化至15%;20min~30min,流动相a的体积百分数由15%变化至17%;30min~40min,流动相a的体积百分数由17%变化至19%;40min~55min,流动相a的体积百分数从19%变化至21%;55min~60min,流动相a的体积百分数从21%变化至23%。
11.在其中一个实施例中,所述磷酸二氢钠水溶液的ph值为2.8~3.2。
12.在其中一个实施例中,超高效液相色谱分析的条件还包括:色谱柱为十八烷基硅烷键合硅胶柱;柱温为25℃~29℃;流速为0.26ml/min~0.4ml/min;波长为200nm~210nm。
13.在其中一个实施例中,制备苍耳子中药配方颗粒供试品溶液或炒苍耳子中药配方颗粒供试品溶液包括如下步骤:
14.将所述苍耳子中药配方颗粒或所述炒苍耳子中药配方颗粒研磨后与体积分数为30%~50%的甲醇水溶液混合,超声提取,然后过滤,取滤液。
15.在其中一个实施例中,制备第一混合对照品溶液包括如下步骤:
16.将原儿茶酸、新绿原酸、绿原酸、隐绿原酸、咖啡酸、1,3-o-二咖啡酰奎宁酸、3,5-o-二咖啡酰奎宁酸、4,5-o-二咖啡酰奎宁酸对照品与体积分数为40%~60%的甲醇水溶液混合;
17.制备第二混合对照品溶液包括如下步骤:
18.将羧基苍术苷三钾盐、苍术苷二钾盐对照品与体积分数为5%~15%的甲醇水溶液混合。
19.在其中一个实施例中,所述苍耳子中药配方颗粒或所述炒苍耳子中药配方颗粒的指纹图谱包含24个共有峰;其中,以峰16为参照峰,峰1的相对保留时间为0.267
±
10%,峰2的相对保留时间为0.291
±
10%,峰3的相对保留时间为0.307
±
10%,峰4的相对保留时间为0.335
±
10%,峰5的相对保留时间为0.411
±
10%,峰6的相对保留时间为0.424
±
10%,峰7的相对保留时间为0.454
±
10%,峰9的相对保留时间为0.53
±
10%,峰11的相对保留时间为0.665
±
10%,峰15的相对保留时间为0.946
±
10%,峰17的相对保留时间为1.122
±
10%,峰18的相对保留时间为1.289
±
10%,峰19的相对保留时间为1.61
±
10%,峰20的相对保留时间为1.703
±
10%。
20.在其中一个实施例中,所述苍耳子中药配方颗粒或所述炒苍耳子中药配方颗粒的指纹图谱中,峰8为原儿茶酸的色谱峰,峰10为新绿原酸的色谱峰,峰12为绿原酸的色谱峰,峰13为隐绿原酸的色谱峰,峰14为咖啡酸的色谱峰,峰16为1,3-o-二咖啡酰奎宁酸的色谱峰,峰21为羧基苍术苷的色谱峰,峰22为3,5-o-二咖啡酰奎宁酸的色谱峰,峰23为4,5-o-二咖啡酰奎宁酸的色谱峰,峰24为苍术苷的色谱峰。
21.本发明还提供一种苍耳子中药配方颗粒与炒苍耳子中药配方颗粒的鉴别方法,包括如下步骤:
22.获取如上所述的指纹图谱构建方法所构建的苍耳子中药配方颗粒的指纹图谱与炒苍耳子中药配方颗粒的指纹图谱,采用正交偏最小二乘判别分析法建立判别分析模型;
23.取待测物,进行超高效液相色谱分析,将获得的所述待测物的图谱导入所述判别分析模型中,获得预测值y
pred
;0.807≤y
pred
≤1.177,所述待测物为苍耳子中药配方颗粒;或-0.198≤y
pred
≤0.215,所述待测物为炒苍耳子中药配方颗粒;
24.其中,超高效液相色谱分析的条件包括:流动相a为乙腈,流动相b为磷酸二氢钠水溶液;采用梯度洗脱;所述梯度洗脱的程序包括:0min~10min,流动相a的体积百分数从2.5%变化至10%;10min~20min,流动相a的体积百分数从10%变化至15%;20min~30min,流动相a的体积百分数由15%变化至17%;30min~40min,流动相a的体积百分数由17%变化至19%;40min~55min,流动相a的体积百分数从19%变化至21%;55min~60min,流动相a的体积百分数从21%变化至23%。
25.在其中一个实施例中,建立判别分析模型包括如下步骤:
26.以1,3-o-二咖啡酰奎宁酸的色谱峰为参照峰,分别计算所述苍耳子中药配方颗粒的指纹图谱中24个共有峰的相对峰面积与所述炒苍耳子中药配方颗粒的指纹图谱中24个共有峰的相对峰面积,然后导入多元统计分析软件,采用正交偏最小二乘判别分析法进行拟合。
27.在其中一个实施例中,所述磷酸二氢钠水溶液的ph值为2.8~3.2。
28.在其中一个实施例中,超高效液相色谱分析的条件还包括:色谱柱为十八烷基硅烷键合硅胶柱;柱温为25℃~29℃;流速为0.26ml/min~0.4ml/min;波长为200nm~210nm。
29.本发明还提供一种苍耳子中药配方颗粒或炒苍耳子中药配方颗粒的检测方法,包括如下步骤:
30.取苍耳子中药配方颗粒或炒苍耳子中药配方颗粒,制备待测溶液;
31.将所述待测溶液进行超高效液相色谱分析;
32.其中,超高效液相色谱分析的条件包括:流动相a为乙腈,流动相b为磷酸二氢钠水溶液;采用梯度洗脱;所述梯度洗脱的程序包括:0min~10min,流动相a的体积百分数从2.5%变化至10%;10min~20min,流动相a的体积百分数从10%变化至15%;20min~30min,流动相a的体积百分数由15%变化至17%;30min~40min,流动相a的体积百分数由17%变化至19%;40min~55min,流动相a的体积百分数从19%变化至21%;55min~60min,流动相a的体积百分数从21%变化至23%。
33.在其中一个实施例中,所述磷酸二氢钠水溶液的ph值为2.8~3.2。
34.在其中一个实施例中,超高效液相色谱分析的条件还包括:色谱柱为十八烷基硅烷键合硅胶柱;柱温为25℃~29℃;流速为0.26ml/min~0.4ml/min;波长为200nm~210nm。
35.与现有技术相比较,本发明的苍耳子中药配方颗粒或炒苍耳子中药配方颗粒的指纹图谱构建方法具有如下有益效果:
36.本发明采用超高效液相色谱法建立苍耳子中药配方颗粒与炒苍耳子中药配方颗粒的指纹图谱,通过筛选流动相分别为乙腈与磷酸二氢钠水溶液,并限定洗脱方式,最终构建的苍耳子与炒苍耳子中药配方颗粒的指纹图谱的指纹图谱可同时表征酚酸类成分与水溶性苷类成分,能够全面地反映苍耳子与炒苍耳子中药配方颗粒的主要化学成分信息,色谱峰分离度良好,基线平稳,具有耗时短、操作简单、特征性强、专属性与重现性好的优势。
37.进一步地,本发明的苍耳子中药配方颗粒与炒苍耳子中药配方颗粒的指纹图谱构建方法可以结合正交偏最小二乘法建立判别分析模型,达到准确鉴别苍耳子与炒苍耳子中
药配方颗粒的目的,从而保证临床用药的安全性和有效性。
附图说明
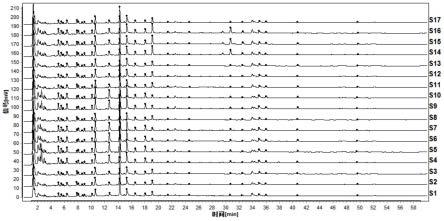
38.图1为本发明提供的苍耳子中药配方颗粒的uplc图谱;
39.图2为本发明提供的炒苍耳子中药配方颗粒的uplc图谱;
40.图3为本发明提供的苍耳子、炒苍耳子中药配方颗粒与对照品的对比图谱;
41.图4为本发明提供的不同色谱柱的对比图谱;
42.图5为本发明提供的不同梯度洗脱程序的对比图谱;
43.图6为本发明提供的不同流动相ph值的对比图谱;
44.图7为本发明提供的不同柱温的对比图谱;
45.图8为本发明提供的不同流速的对比图谱;
46.图9为本发明提供的正交偏最小二乘法判别分析模型得分图。
具体实施方式
47.为了便于理解本发明,下面将参照相关附图对本发明进行更全面的描述。附图中给出了本发明的较佳实施方式。但是,本发明可以以许多不同的形式来实现,并不限于本文所描述的实施方式。相反地,提供这些实施方式的目的是使对本发明的公开内容的理解更加透彻全面。
48.此外,术语“第一”、“第二”仅用于描述目的,而不能理解为指示或暗示相对重要性或者隐含指明所指示的技术特征的数量。由此,限定有“第一”、“第二”的特征可以明示或者隐含地包括至少一个该特征。在发明的描述中,“多个”的含义是至少两个,例如两个,三个等,除非另有明确具体的限定。在本发明的描述中,“若干”的含义是至少一个,例如一个,两个等,除非另有明确具体的限定。
49.本发明中的词语“优选地”、“更优选地”等是指,在某些情况下可提供某些有益效果的本发明实施方案。然而,在相同的情况下或其他情况下,其他实施方案也可能是优选的。此外,对一个或多个优选实施方案的表述并不暗示其他实施方案不可用,也并非旨在将其他实施方案排除在本发明的范围之外。
50.当本文中公开一个数值范围时,上述范围视为连续,且包括该范围的最小值及最大值,以及这种最小值与最大值之间的每一个值。进一步地,当范围是指整数时,包括该范围的最小值与最大值之间的每一个整数。此外,当提供多个范围描述特征或特性时,可以合并该范围。换言之,除非另有指明,否则本文中所公开之所有范围应理解为包括其中所归入的任何及所有的子范围。
51.除非另外指明,所有百分比、分数和比率都是按本发明组合物的总质量计算的。除非另外指明,有关所列成分的所有质量均给予活性物质的含量,因此它们不包括在可商购获得的材料中可能包含的溶剂或副产物。本文术语“质量百分比含量”可用符号“%”表示。除非另外指明,在本文中所有的分子量都是以道尔顿为单位表示的重均分子量。除非另外指明,在本文中所有配制和测试发生在25℃的环境。本文中“包括”、“包含”、“含”、“含有”、“具有”或其它变体意在涵盖非封闭式包括,这些术语之间不作区分。术语“包含”是指可加入不影响最终结果的其它步骤和成分。本发明的组合物和方法/工艺包含、由其组成和基本
上由本文描述的必要元素和限制项以及本文描述的任一的附加的或任选的成分、组份、步骤或限制项组成。本文中术语“效能”、“性能”、“效果”、“功效”之间不作区分。
52.除非另有定义,本文所使用的所有的技术和科学术语与属于本发明的技术领域的技术人员通常理解的含义相同。本文中在本发明的说明书中所使用的术语只是为了描述具体的实施方式的目的,不是旨在于限制本发明。本文所使用的术语“和/或”包括一个或多个相关的所列项目的任意的和所有的组合。
53.本发明提供了一种苍耳子中药配方颗粒或炒苍耳子中药配方颗粒的指纹图谱构建方法,包括如下步骤:
54.取原儿茶酸、新绿原酸、绿原酸、隐绿原酸、咖啡酸、1,3-o-二咖啡酰奎宁酸、3,5-o-二咖啡酰奎宁酸与4,5-o-二咖啡酰奎宁酸对照品,制备第一混合对照品溶液;取羧基苍术苷三钾盐与苍术苷二钾盐对照品,制备第二混合对照品溶液;
55.取苍耳子中药配方颗粒或炒苍耳子中药配方颗粒,制备苍耳子中药配方颗粒供试品溶液或炒苍耳子中药配方颗粒供试品溶液;
56.分别将苍耳子中药配方颗粒供试品溶液或炒苍耳子中药配方颗粒供试品溶液、第一混合对照品溶液与第二混合对照品溶液进行超高效液相色谱分析,构建苍耳子中药配方颗粒的指纹图谱或炒苍耳子中药配方颗粒的指纹图谱;
57.其中,超高效液相色谱分析的条件包括:流动相a为乙腈,流动相b为磷酸二氢钠水溶液;采用梯度洗脱;梯度洗脱的程序包括:0min~10min,流动相a的体积百分数从2.5%变化至10%;10min~20min,流动相a的体积百分数从10%变化至15%;20min~30min,流动相a的体积百分数由15%变化至17%;30min~40min,流动相a的体积百分数由17%变化至19%;40min~55min,流动相a的体积百分数从19%变化至21%;55min~60min,流动相a的体积百分数从21%变化至23%。
58.在一个具体的示例中,磷酸二氢钠水溶液的ph值为2.8~3.2。
59.可以理解地,在本发明中,磷酸二氢钠水溶液的ph值包括但不限于2.8、2.9、3、3.1、3.2。优选地,磷酸二氢钠水溶液的ph值为3。
60.在一个具体的示例中,超高效液相色谱分析的条件还包括:色谱柱为十八烷基硅烷键合硅胶柱;柱温为25℃~29℃;流速为0.26ml/min~0.4ml/min;波长为200nm~210nm。
61.可以理解地,在本发明中,柱温包括但不限于25℃、26℃、27℃、28℃、29℃;流速包括但不限于0.26ml/min、0.27ml/min、0.28ml/min、0.29ml/min、0.3ml/min、0.31ml/min、0.32ml/min、0.33ml/min、0.34ml/min、0.35ml/min、0.36ml/min、0.37ml/min、0.38ml/min、0.39ml/min、0.4ml/min;波长包括但不限于200nm、201nm、202nm、203nm、204nm、205nm、206nm、207nm、208nm、209nm、210nm。
62.在一个具体的示例中,色谱柱为acquity hss t3色谱柱。
63.在一个具体的示例中,制备苍耳子中药配方颗粒供试品溶液或炒苍耳子中药配方颗粒供试品溶液包括如下步骤:
64.将苍耳子中药配方颗粒或炒苍耳子中药配方颗粒研磨后与体积分数为30%~50%的甲醇水溶液混合,超声提取,然后过滤,取滤液。
65.可以理解地,在本发明中,甲醇水溶液的体积分数包括但不限于30%、32%、34%、36%、38%、39%、40%、41%、42%、43%、44%、45%、46%、48%、50%。
66.更具体地,制备苍耳子中药配方颗粒供试品溶液或炒苍耳子中药配方颗粒供试品溶液包括如下步骤:
67.分别取苍耳子中药配方颗粒或炒苍耳子中药配方颗粒,研细,加入50%甲醇水溶液25ml,称定重量,超声提取,放冷,用50%甲醇溶液补足减失的重量,摇匀,滤过,取续滤液。
68.更具体地,超声提取的条件包括:功率为400w,频率为40khz,时间为30分钟。
69.一个具体的示例中,制备第一混合对照品溶液包括如下步骤:
70.将原儿茶酸、新绿原酸、绿原酸、隐绿原酸、咖啡酸、1,3-o-二咖啡酰奎宁酸、3,5-o-二咖啡酰奎宁酸、4,5-o-二咖啡酰奎宁酸对照品与体积分数为40%~60%的甲醇水溶液混合。
71.在一个具体的示例中,制备第二混合对照品溶液包括如下步骤:
72.将羧基苍术苷三钾盐、苍术苷二钾盐对照品与体积分数为5%~15%的甲醇水溶液混合。
73.在一个具体的示例中,苍耳子中药配方颗粒或炒苍耳子中药配方颗粒的指纹图谱包含24个共有峰;其中,以峰16为参照峰,峰1的相对保留时间为0.267
±
10%,峰2的相对保留时间为0.291
±
10%,峰3的相对保留时间为0.307
±
10%,峰4的相对保留时间为0.335
±
10%,峰5的相对保留时间为0.411
±
10%,峰6的相对保留时间为0.424
±
10%,峰7的相对保留时间为0.454
±
10%,峰9的相对保留时间为0.53
±
10%,峰11的相对保留时间为0.665
±
10%,峰15的相对保留时间为0.946
±
10%,峰17的相对保留时间为1.122
±
10%,峰18的相对保留时间为1.289
±
10%,峰19的相对保留时间为1.61
±
10%,峰20的相对保留时间为1.703
±
10%。
74.在一个具体的示例中,苍耳子中药配方颗粒或所述炒苍耳子中药配方颗粒的指纹图谱中,峰8为原儿茶酸的色谱峰,峰10为新绿原酸的色谱峰,峰12为绿原酸的色谱峰,峰13为隐绿原酸的色谱峰,峰14为咖啡酸的色谱峰,峰16为1,3-o-二咖啡酰奎宁酸的色谱峰,峰21为羧基苍术苷的色谱峰,峰22为3,5-o-二咖啡酰奎宁酸的色谱峰,峰23为4,5-o-二咖啡酰奎宁酸的色谱峰,峰24为苍术苷的色谱峰。
75.本发明还提供一种苍耳子中药配方颗粒与炒苍耳子中药配方颗粒的鉴别方法,包括如下步骤:
76.获取上述指纹图谱构建方法所构建的苍耳子中药配方颗粒的指纹图谱与炒苍耳子中药配方颗粒的指纹图谱,采用正交偏最小二乘判别分析法建立判别分析模型;
77.取待测物,进行超高效液相色谱分析,将获得的待测物的图谱导入判别分析模型中,获得预测值y
pred
;0.807≤y
pred
≤1.177,待测物为苍耳子中药配方颗粒;或-0.198≤y
pred
≤0.215,待测物为炒苍耳子中药配方颗粒;
78.其中,超高效液相色谱分析的条件包括:流动相a为乙腈,流动相b为磷酸二氢钠水溶液;采用梯度洗脱;梯度洗脱的程序包括:0min~10min,流动相a的体积百分数从2.5%变化至10%;10min~20min,流动相a的体积百分数从10%变化至15%;20min~30min,流动相a的体积百分数由15%变化至17%;30min~40min,流动相a的体积百分数由17%变化至19%;40min~55min,流动相a的体积百分数从19%变化至21%;55min~60min,流动相a的体积百分数从21%变化至23%。
79.可以理解地,在本发明中,当y
pred
在上述两个范围以外,则不属于该模型的判定范围内,判定结果无效。
80.在一个具体的示例中,建立判别分析模型包括如下步骤:
81.以1,3-o-二咖啡酰奎宁酸的色谱峰为参照峰,分别计算苍耳子中药配方颗粒的指纹图谱中24个共有峰的相对峰面积与炒苍耳子中药配方颗粒的指纹图谱中24个共有峰的相对峰面积,然后导入多元统计分析软件,采用正交偏最小二乘判别分析法进行拟合。
82.在一个具体的示例中,判别分析模型的拟合参数r2y大于0.9,判别分析模型的预测参数q2y大于0.9。
83.在一个具体的示例中,磷酸二氢钠水溶液的ph值为2.8~3.2。
84.可以理解地,在本发明中,磷酸二氢钠水溶液的ph值包括但不限于2.8、2.9、3、3.1、3.2。优选地,磷酸二氢钠水溶液的ph值为3。
85.在一个具体的示例中,超高效液相色谱分析的条件还包括:色谱柱为十八烷基硅烷键合硅胶柱;柱温为25℃~29℃;流速为0.26ml/min~0.4ml/min;波长为200nm~210nm。
86.可以理解地,在本发明中,柱温包括但不限于25℃、26℃、27℃、28℃、29℃;流速包括但不限于0.26ml/min、0.27ml/min、0.28ml/min、0.29ml/min、0.3ml/min、0.31ml/min、0.32ml/min、0.33ml/min、0.34ml/min、0.35ml/min、0.36ml/min、0.37ml/min、0.38ml/min、0.39ml/min、0.4ml/min;波长包括但不限于200nm、201nm、202nm、203nm、204nm、205nm、206nm、207nm、208nm、209nm、210nm。
87.在一个具体的示例中,色谱柱为acquity hss t3色谱柱。
88.本发明还提供一种苍耳子中药配方颗粒或炒苍耳子中药配方颗粒的检测方法,包括如下步骤:
89.取苍耳子中药配方颗粒或炒苍耳子中药配方颗粒,制备待测溶液;
90.将待测溶液进行超高效液相色谱分析;
91.其中,超高效液相色谱分析的条件包括:流动相a为乙腈,流动相b为磷酸二氢钠水溶液;采用梯度洗脱;梯度洗脱的程序包括:0min~10min,流动相a的体积百分数从2.5%变化至10%;10min~20min,流动相a的体积百分数从10%变化至15%;20min~30min,流动相a的体积百分数由15%变化至17%;30min~40min,流动相a的体积百分数由17%变化至19%;40min~55min,流动相a的体积百分数从19%变化至21%;55min~60min,流动相a的体积百分数从21%变化至23%。
92.在一个具体的示例中,磷酸二氢钠水溶液的ph值为2.8~3.2。
93.可以理解地,在本发明中,磷酸二氢钠水溶液的ph值包括但不限于2.8、2.9、3、3.1、3.2。优选地,磷酸二氢钠水溶液的ph值为3。
94.在一个具体的示例中,超高效液相色谱分析的条件还包括:色谱柱为十八烷基硅烷键合硅胶柱;柱温为25℃~29℃;流速为0.26ml/min~0.4ml/min;波长为200nm~210nm。
95.可以理解地,在本发明中,柱温包括但不限于25℃、26℃、27℃、28℃、29℃;流速包括但不限于0.26ml/min、0.27ml/min、0.28ml/min、0.29ml/min、0.3ml/min、0.31ml/min、0.32ml/min、0.33ml/min、0.34ml/min、0.35ml/min、0.36ml/min、0.37ml/min、0.38ml/min、0.39ml/min、0.4ml/min;波长包括但不限于200nm、201nm、202nm、203nm、204nm、205nm、206nm、207nm、208nm、209nm、210nm。
96.在一个具体的示例中,色谱柱为acquity hss t3色谱柱。
97.以下结合具体实施例对本发明的苍耳子中药配方颗粒与炒苍耳子中药配方颗粒的指纹图谱构建与鉴别方法做进一步详细的说明。以下实施例中所用的原料,如无特别说明,均为市售产品。
98.实施例1
99.本实施例提供一种苍耳子中药配方颗粒与炒苍耳子中药配方颗粒的指纹图谱构建方法,具体如下:
100.1仪器和试药
101.1290infinity ii型超高效液相色谱仪(安捷伦科技有限公司);acquity hss t3型色谱柱,柱长为150mm,内径为2.1mm,粒径为1.8μm(沃特世科技有限公司);me204e型、xp26型电子天平(梅特勒托利多科技有限公司);kq-500de型超声仪(昆山市超声仪器有限公司)。
102.绿原酸(批号110753-202119,含量:96.3%)、咖啡酸(批号110885-201703,含量:99.7%)、1,3-o-二咖啡酰奎宁酸(批号111717-201402,含量:94.5%)、3,5-o-二咖啡酰奎宁酸(批号111782-201807,含量:99.5%)、4,5-o-二咖啡酰奎宁酸(批号111894-202103,含量:95.2%)、原儿茶酸(批号110809-201906,含量:97.7%)、羧基苍术苷三钾盐(批号112027-201701,含量:95.1%)、苍术苷二钾盐(批号111989-201801,含量:92.6%)对照品均购于中国食品药品检定研究院;新绿原酸(批号dstdx001503,含量:99.6%)、隐绿原酸(批号dst220104-035,含量:99.3%)对照品均购于成都乐美天医药科技有限公司。乙腈为色谱纯,水为超纯水,其余试剂均为分析纯。
103.取17批苍耳子药材样品(编号:1~17),产地信息见表1。取苍耳子药材,经除去杂质,得苍耳子饮片。取苍耳子饮片,置炒锅内加热炒至表面黄褐色,取出晾凉,去刺并筛净,得炒苍耳子饮片。分别取苍耳子饮片和炒苍耳子饮片,捣碎,加水煎煮,煎液浓缩后干燥,加适量辅料制成配方颗粒,苍耳子中药配方颗粒编号为s1~s17,炒苍耳子中药配方颗粒编号为sk1~sk17。规格统一为:每1g配方颗粒相当于10g饮片。
104.表1苍耳子药材产地信息
[0105][0106]
2苍耳子与炒苍耳子中药配方颗粒的指纹图谱的建立
[0107]
2.1对照品溶液的制备
[0108]
分别取原儿茶酸、新绿原酸、绿原酸、隐绿原酸、咖啡酸、1,3-o-二咖啡酰奎宁酸、3,5-o-二咖啡酰奎宁酸、4,5-o-二咖啡酰奎宁酸对照品适量,精密称定,加50%甲醇水溶液制成每1ml各含50μg的混合对照品溶液a;分别取羧基苍术苷三钾盐、苍术苷二钾盐对照品适量,精密称定,加10%甲醇水溶液制成每1ml各含200μg的混合对照品溶液b。
[0109]
2.2供试品溶液的制备
[0110]
分别取苍耳子中药配方颗粒、炒苍耳子中药配方颗粒适量,研细,取0.2g,精密称定,置具塞锥形瓶中,精密加入50%甲醇水溶液25ml,称定重量,超声提取(功率400w,频率40khz)30分钟,放冷,用50%甲醇溶液补足减失的重量,摇匀,滤过,取续滤液,即得苍耳子中药配方颗粒、炒苍耳子中药配方颗粒供试品溶液。
[0111]
2.3超高效液相色谱条件
[0112]
以可兼容100%水相的十八烷基硅烷键合高强度硅胶颗粒为填充剂,以乙腈为流动相a,0.01mol/l磷酸二氢钠溶液(加磷酸调节ph至3.0)为流动相b,按下表2中的规定程序进行梯度洗脱,流速为每分钟0.3ml,柱温为27℃,检测波长为203nm。
[0113]
分别精密吸取对照品溶液a、对照品溶液b、苍耳子中药配方颗粒与炒苍耳子中药配方颗粒供试品溶液各1μl,注入超高效液相色谱仪,测定,得色谱图。
[0114]
表2梯度洗脱程序
[0115][0116]
2.4 uplc指纹图谱的建立
[0117]
将色谱图导入“中药色谱指纹图谱相似度评价系统软件(2012版本)”软件,见图1和图2。分别以s1、sk1为参照,先进行全谱峰匹配找出共有峰,再选取峰形较好的24个共有峰进行多点校正和mark峰匹配,设置时间窗宽度为0.1,采用中位数法生产相应的对照指纹图谱。分别计算17批苍耳子、炒苍耳子中药配方颗粒指纹图谱与对照指纹图谱的相似度,结果见表3,其相似度均在0.93、0.96以上,表明其指纹图谱的重现性良好。
[0118]
表3指纹图谱相似度
[0119]
[0120][0121]
2.5色谱峰的指认和定位
[0122]
根据已知对照品,共指认了10个色谱峰,分别为:原儿茶酸(峰8)、新绿原酸(峰10)、绿原酸(峰12)、隐绿原酸(峰13)、咖啡酸(峰14)、1,3-o-二咖啡酰奎宁酸(峰16)、羧基苍术苷(峰21)、3,5-o-二咖啡酰奎宁酸(峰22)、4,5-o-二咖啡酰奎宁酸(峰23)、苍术苷(峰24),结果见图3,保留时间见表4。已指认的10个色谱峰可以以对照品的保留时间来定位。以1,3-o-二咖啡酰奎宁酸色谱峰为参照峰s,计算峰1~峰24的相对保留时间,结果见表5、表6。剩余的14个色谱峰可以通过相对保留时间来定位,规定其相对保留时间应在规定值的
±
10%范围之内,规定值为:0.267(峰1)、0.291(峰2)、0.307(峰3)、0.335(峰4)、0.411(峰5)、0.424(峰6)、0.454(峰7)、0.530(峰9)、0.665(峰11)、0.946(峰15)、1.122(峰17)、1.289(峰18)、1.610(峰19)、1.703(峰20)。
[0123]
表4 24个共有峰的保留时间及对照品指认结果
[0124][0125]
表5相对保留时间
[0126]
[0127][0128]
表6相对保留时间
[0129]
[0130][0131]
3色谱条件的建立与优化过程
[0132]
羧基苍术苷和苍术苷属于紫外末端吸收,最大吸收波长约为203nm,经过实验摸索,优选检测波长为203nm,所得色谱图中的峰信息比较丰富。甲醇的紫外吸收波长在200nm左右,而乙腈在190nm左右,为减轻流动相对检测基线的干扰,因此优选乙腈为流动相。甲酸和乙酸含有羧基官能团在200nm左右有吸收,存在干扰,而磷酸不存在这样的结构,因此优选用磷酸和磷酸二氢钠缓冲盐溶液。
[0133]
3.1色谱柱的选择
[0134]
取苍耳子中药配方颗粒供试品溶液,分别采用以苯基键合硅胶颗粒、十八烷基硅烷键合高强度硅胶颗粒为固定相的色谱柱,以乙腈为流动相a,0.01mol/l磷酸二氢钠溶液(加磷酸调节ph至3.0)为流动相b,按表2中的规定比例进行梯度洗脱,流速为每分钟0.3ml,柱温为27℃,检测波长为203nm,得色谱图,见图4。
[0135]
结果显示:苯基键合硅胶色谱柱所得色谱图中的峰信息较少,不能全面反应苍耳子的整体特征;而十八烷基硅烷键合高强度硅胶色谱柱中的峰信息量比较丰富。因为流动相初始比例为高比例水相,所以色谱柱选用可以兼容100%水相的以十八烷基硅烷键合高强度硅胶为固定相的acquity hss t3色谱柱。
[0136]
3.2梯度洗脱程序的考察
[0137]
梯度洗脱程序是影响色谱峰分离度的主要因素之一,因此考察不同梯度洗脱程序对色谱图的影响。取苍耳子中药配方颗粒供试品溶液,采用acquity hss t3色谱柱,以乙腈为流动相a,以0.01mol/l磷酸二氢钠溶液(加磷酸调节ph至3.0)为流动相b,按表7中的规定的比例进行梯度洗脱,流速为每分钟0.3ml,柱温为27℃,检测波长为203nm,得色谱图,见图5。
[0138]
表7不同梯度洗脱比例
[0139][0140]
结果显示:采用梯度洗脱程序a时,所得色谱图的基线不平稳且无法获得24个关键色谱峰。经过大量实验优化后得到梯度洗脱程序b,在该梯度洗脱程序下,所得的色谱图基线平稳且24个关键色谱峰峰形较好,分离度较好,因此采用梯度洗脱程序b。
[0141]
3.3流动相b的ph值考察
[0142]
流动相ph值是影响色谱峰分离度的主要因素之一,因此考察流动相b的不同ph值对色谱图的影响。取苍耳子中药配方颗粒供试品溶液,采用acquity hss t3色谱柱,以乙腈为流动相a,分别以0.01mol/l磷酸二氢钠溶液(加磷酸调节ph至2.5)、0.01mol/l磷酸二氢钠溶液(加磷酸调节ph至3.0)、0.01mol/l磷酸二氢钠溶液(加磷酸调节ph至3.5)为流动相b,按表2中的规定比例进行梯度洗脱,流速为每分钟0.3ml,柱温为27℃,检测波长为203nm,得色谱图,见图6。
[0143]
结果显示:流动相b的ph值采用2.5或3.5时,色谱图中的关键峰峰形欠佳,分离度较差,不能完整得到24个关键色谱峰。而流动相b的ph值调节至3.0时,所得的色谱图的24个关键色谱峰峰形较好,分离度较好,因此优选流动相b的ph值为3.0。
[0144]
3.4色谱柱温度考察
[0145]
取苍耳子中药配方颗粒供试品溶液,采用acquity hss t3色谱柱,以乙腈为流动相a,以0.01mol/l磷酸二氢钠溶液(加磷酸调节ph至3.0)为流动相b,按表2中的规定比例进行梯度洗脱,流速为每分钟0.3ml,分别设置柱温为25℃、26℃、27℃、28℃、29℃,检测波长为203nm,得色谱图,见图7。
[0146]
结果显示:在上述柱温中,各色谱峰峰形较好,分离度较好,故确定柱温范围为25~29℃。
[0147]
3.5流速考察
[0148]
取苍耳子中药配方颗粒供试品溶液,采用acquity hss t3色谱柱,以乙腈为流动相a,以0.01mol/l磷酸二氢钠溶液(加磷酸调节ph至3.0)为流动相b,按表2中的规定比例进行梯度洗脱,分别设置流速为每分钟0.26ml、0.30ml、0.35ml、0.40ml,设置柱温为27℃,检测波长为203nm,得色谱图,见图8。
[0149]
结果显示,流速范围在每分钟0.26~0.40ml,各色谱峰峰形较好,分离度较好,故确定流速为0.26~0.40ml/min。
[0150]
综上所述,色谱条件为:选用以十八烷基硅烷键合高强度硅胶颗粒为固定相的色谱柱,以乙腈为流动相a,0.01mol/l磷酸二氢钠溶液(加磷酸调节ph至3.0)为流动相b,色谱柱温度范围可为25~29℃,流动相流速范围可为0.26~0.40ml/min。采用梯度洗脱方式,洗脱程序为:0~10min,流动相a的体积百分比由2.5%上升至10%;10~20min,流动相a的体积百分比由10%上升至15%;20~30min,流动相a的体积百分比由15%上升至17%;30~
40min,流动相a的体积百分比由17%上升至19%;40~55min,流动相a的体积百分比由19%上升至21%;55~60min,流动相a的体积百分比由21%上升至23%;检测波长为203nm。
[0151]
优选的色谱条件为:以可兼容100%水相的十八烷基硅烷键合高强度硅胶颗粒为固定相的色谱柱,以乙腈为流动相a,0.01mol/l磷酸二氢钠溶液(加磷酸调节ph至3.0)为流动相b,色谱柱温度为27℃,流动相流速为0.30ml/min。采用梯度洗脱方式,洗脱程序为:0~10min,流动相a的体积百分比由2.5%上升至10%;10~20min,流动相a的体积百分比由10%上升至15%;20~30min,流动相a的体积百分比由15%上升至17%;30~40min,流动相a的体积百分比由17%上升至19%;40~55min,流动相a的体积百分比由19%上升至21%;55~60min,流动相a的体积百分比由21%上升至23%;检测波长为203nm。
[0152]
4供试品溶液的制备考察
[0153]
4.1提取溶剂考察
[0154]
分别取苍耳子中药配方颗粒适量,研细,取0.2g,精密称定,置具塞锥形瓶中,分别精密加入30%甲醇水溶液、40%甲醇水溶液、50%甲醇水溶液25ml,称定重量,超声提取(功率400w,频率40khz)30分钟,放冷,分别用提取溶剂补足减失的重量,摇匀,滤过,取续滤液,即得供试品溶液。按上述优选的色谱条件检测,以24个共有峰的总峰面积为评价指标,考察提取溶剂,结果见表8。
[0155]
表8不同提取溶剂所得总峰面积
[0156][0157]
结果表明,30%、40%、50%甲醇水溶液提取效果无明显差别,故确定提取溶剂可为30%~50%甲醇水溶液。
[0158]
4.2提取方式考察
[0159]
分别取苍耳子中药配方颗粒适量,研细,取0.2g,精密称定,置具塞锥形瓶中,分别精密加入50%甲醇水溶液25ml,称定重量,分别采用加热回流和超声提取(功率400w,频率40khz)30分钟,放冷,分别用提取溶剂补足减失的重量,摇匀,滤过,取续滤液,即得供试品溶液。按上述优选的色谱条件检测,以总峰面积为评价指标,考察提取方式,结果见表9。
[0160]
表9不同提取方式所得总峰面积
[0161][0162]
结果表明,超声提取效果较好,故确定提取方式为超声提取。
[0163]
4.3提取溶剂用量考察
[0164]
分别取苍耳子中药配方颗粒适量,研细,取0.2g,精密称定,置具塞锥形瓶中,分别精密加入10ml、25ml、50ml的50%甲醇水溶液,称定重量,超声提取(功率400w,频率40khz)30分钟,放冷,分别用提取溶剂补足减失的重量,摇匀,滤过,取续滤液,即得供试品溶液。按
上述优选的色谱条件检测,以总峰面积为评价指标,考察提取溶剂用量,结果见表10。
[0165]
表10不同提取溶剂用量所得总峰面积
[0166][0167]
结果表明,25ml和50ml溶剂量的提取效果接近,基于节约试剂考虑,故确定提取溶剂为25ml。
[0168]
4.4提取时间考察
[0169]
分别取苍耳子中药配方颗粒适量,研细,取0.2g,精密称定,置具塞锥形瓶中,分别精密加入50%甲醇水溶液25ml,称定重量,分别超声提取(功率400w,频率40khz)20min、30min、40min,放冷,分别用提取溶剂补足减失的重量,摇匀,滤过,取续滤液,即得供试品溶液。按上述优选的色谱条件检测,以总峰面积为评价指标,考察提取溶剂用量,结果见表11。
[0170]
表11不同提取时间所得总峰面积
[0171][0172][0173]
结果表明,超声20~30min后再增加提取时间对总峰面积增加效果不明显,故超声提取时间可为20~30min。
[0174]
综上所述,供试品的制备条件可为:取苍耳子中药配方颗粒,研细,取0.2g,精密称定,置具塞锥形瓶中,精密加入30~50%甲醇水溶液25ml,称定重量,超声提取(功率400w,频率40khz)20~30分钟,放冷,用提取溶剂补足减失的重量,摇匀,滤过,取续滤液,即得供试品溶液。
[0175]
优选的供试品制备条件为:取苍耳子中药配方颗粒,研细,取0.2g,精密称定,置具塞锥形瓶中,精密加入50%甲醇水溶液25ml,称定重量,超声提取(功率400w,频率40khz)30分钟,放冷,用提取溶剂补足减失的重量,摇匀,滤过,取续滤液,即得供试品溶液。
[0176]
5方法学考察
[0177]
5.1精密度试验取苍耳子中药配方颗粒样品,按2.2项下的供试品制备方法制备1份供试品溶液,按2.3项下的色谱条件重复进样测定6次。以1,3-o-二咖啡酰奎宁酸色谱峰为参照峰s,分别计算24个共有峰与s峰的相对保留时间和相对峰面积,并计算rsd值。结果显示,各峰相对保留时间和相对峰面积的rsd均小于3.0%(n=6),说明该仪器精密度良好。
[0178]
5.2稳定性试验取苍耳子中药配方颗粒样品,按2.2项下的供试品制备方法制备6份供试品溶液,按按2.3项下的色谱条件分别在0、2、4、8、12、24h进样测定。以1,3-o-二咖啡酰奎宁酸色谱峰为参照峰s,24个共有峰相对保留时间的rsd均小于2.0%(n=6),相对峰面积的rsd均小于3.0%(n=6),说明供试品溶液在24h内稳定性较好。
[0179]
5.3重复性试验取苍耳子中药配方颗粒样品6份,按2.2项下的供试品制备方法平行制备6份供试品溶液,按2.3项下的色谱条件进样测定。以1,3-o-二咖啡酰奎宁酸色谱峰为参照峰s,24个共有峰相对保留时间的rsd均小于2.0%(n=6),相对峰面积的rsd均小于
3.0%(n=6),表明该方法重复性良好。
[0180]
实施例2
[0181]
本实施例提供一种苍耳子中药配方颗粒与炒苍耳子中药配方颗粒的鉴别方法,具体如下:
[0182]
1正交偏最小二乘法判别分析模型的建立
[0183]
正交偏最小二乘法判别分析模型(opls-da)是一种基于正交偏最小二乘法(orthogonal partial least squares,opls)的有监督模式识别方法,可以对多个样本同时进行多元线性回归以及多组变量之间的相关性分析。正交偏最小二乘回归法是对多元线性回归模型的拓展。其最简单形式为一个描述因变量y和自变量x之间的线性模型:
[0184]
y=β0+β1x1+β2x2+
···
+β
p
x
p
[0185]
其中β0为截距,βi为自变量xi的回归系数。
[0186]
opls-da的基本模型为:
[0187]
x=tp
t
+e
[0188]
y=uq
t
+f
[0189]
其中x(n
×
m)为自变量矩阵,y(n
×
p)为因变量矩阵,t和u分别为x的潜变量得分矩阵以及y的潜变量得分矩阵;p(m
×
f)和q(p
×
f)分别为正交的载荷矩阵;e和f分别为拟合残差矩阵。其中x和y以确保t和u协方差最大化的原则进行分解。模型的质量评价参数包括模型拟合参数r2y以及预测参数q2y:
[0190]
r2y=1-∑(x^-x)2/∑x2[0191]
q2y=1-∑(y^pred-y)2/∑y2[0192]
苍耳子与炒苍耳子中药配方颗粒中的24个共有峰,以1,3-o-二咖啡酰奎宁酸为参照峰,计算各共有峰的相对峰面积,数据见表12和表13。将上述相对峰面积导入计算机统计分析软件simca软件,采用正交偏最小二乘法自动拟合opls-da模型。opls-da模型拟合参数r2y=0.984,预测参数q2y=0.956,均大于0.9,表明所建模型比较稳定且预测能力较强,模型得分图见图9。
[0193]
表12 24个共有峰的相对峰面积
[0194]
[0195][0196]
表13 24个共有峰的相对峰面积
[0197][0198][0199]
从图9可看出:苍耳子中药配方颗粒与炒苍耳子中药配方颗粒可被t[1]轴分为两
大类,表明该模型能够区分苍耳子中药配方颗粒与炒苍耳子中药配方颗粒。对该模型进行排列测试分析,未发现过拟合现象,表明该模型预测能力较好。计算并导出该模型的预测值y
pred
,见表14。
[0200]
表14 opls-da模型预测值y pred
[0201][0202]
结果显示,17批苍耳子中药配方颗粒的y
pred
范围为:0.878~1.143,17批炒苍耳子中药配方颗粒的y
pred
范围为:-0.135~0.118。苍耳子中药配方颗粒的y
pred
控制范围可拟定为平均值
±
3倍sd范围:0.807~1.177,炒苍耳子中药配方颗粒的y
pred
控制范围可拟定为平均值
±
3倍sd范围:-0.198~0.215。该opls-da模型的具体判别标准分三种情况:(1)当0.807≤y
pred
≤1.177时,判定样品属于苍耳子中药配方颗粒;(2)当-0.198≤y
pred
≤0.215时,判定样品属于炒苍耳子中药配方颗粒;(3)当y
pred
在以上两个范围以外,则不属于该模型的判定范围内,判定结果无效。
[0203]
2 opls-da模型的验证
[0204]
按实施例一中2.2项下的供试品制备方法分别制备另外三批苍耳子中药配方颗粒与三批炒苍耳子中药配方颗粒供试品溶液,按实施例一中2.3项下的超高效液相色谱法采集uplc指纹图谱,24个共有峰的相对峰面积见表15与表16,将相对峰面积导入上述判别分析模型,输出预测值y
pred
,结果见表17。
[0205]
表15相对峰面积
[0206][0207]
表16相对峰面积
[0208][0209]
表17预测值y
pred
与判别结果
[0210][0211]
结果表明,根据判别分析模型输出的y
pred
值,可以准确判别苍耳子中药配方颗粒与炒苍耳子中药配方颗粒。
[0212]
3对未知样品的判别分析
[0213]
分别取另外若干批苍耳子中药配方颗粒与若干批炒苍耳子中药配方颗粒共6批样品,遮盖标签品名,当作未知样品,打乱顺序,再随机重新编号为y1~y6。各取适量样品,按实施例一中2.2项下的供试品制备方法制备供试品溶液,按实施例一中2.3项下的超高效液相色谱法采集uplc指纹图谱,24个共有峰的相对峰面积见表18与表19,将相对峰面积导入上述判别分析模型,输出预测值y
pred
,得出判别结果,再与原来的标签品名对照,结果见表20。
[0214]
表18相对峰面积
[0215][0216]
表19相对峰面积
[0217][0218]
表20预测值y
pred
与判别结果
[0219][0220]
结果表明,该判别分析模型可以准确判别苍耳子中药配方颗粒与炒苍耳子中药配
方颗粒。
[0221]
以上所述实施例的各技术特征可以进行任意的组合,为使描述简洁,未对上述实施例中的各个技术特征所有可能的组合都进行描述,然而,只要这些技术特征的组合不存在矛盾,都应当认为是本说明书记载的范围。
[0222]
以上所述实施例仅表达了本发明的几种实施方式,便于具体和详细地理解本发明的技术方案,但并不能因此而理解为对发明专利保护范围的限制。应当指出的是,对于本领域的普通技术人员来说,在不脱离本发明构思的前提下,还可以做出若干变形和改进,这些都属于本发明的保护范围。应当理解,本领域技术人员在本发明提供的技术方案的基础上,通过合乎逻辑的分析、推理或者有限的试验得到的技术方案,均在本发明所附权利要求的保护范围内。因此,本发明专利的保护范围应以所附权利要求的内容为准,说明书及附图可以用于解释权利要求的内容。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1