高效液相色谱法测定司来帕格原料药中有关物质的方法与流程
1.本发明属于药物分析技术领域,具体而言,本发明涉及一种利用高效液相色谱法测定司来帕格原料药中有关物质的方法。
背景技术:
2.司来帕格(selexipag)的化学名称为2-[4-[n-(5,6-二苯基吡嗪-2-基)-n-异丙基氨基]丁氧基]-n-(甲磺酰基)乙酰胺,结构式如下,其是一种口服有效、高选择性和长效的非前列腺素类前列环素受体激动剂,可松弛血管壁平滑肌、扩张血管、降低肺动脉压力,用于治疗成人肺动脉高压,具有选择性高、作用时间长、安全和耐受性等优点。
[0003][0004]
司来帕格原料药中可能存在合成原料和副产物以及降解杂质,已知的杂质有:
[0005]
杂质1(原料杂质):5-氯-2,3-二苯基吡嗪;
[0006]
杂质2(中间体):4-(n-(5,6-二苯基吡嗪-2-基)-n-异丙基氨基)-1-丁醇;
[0007]
杂质3(中间体):2-(4-(n-(5,6-二苯基吡嗪-2-基)-n-异丙基氨基)丁氧基)乙酸叔丁酯;
[0008]
杂质4(中间体及降解产物):2-(4-(n-(5,6-二苯基吡嗪-2-基)-n-异丙基氨基)丁氧基)乙酸;
[0009]
杂质5(副产物):5-(异丙基(4-(2-(甲基磺酰胺基)-2-氧代乙氧基)丁基)氨基)-2,3-二苯基吡嗪1-氧化物;
[0010]
杂质6(副产物):5-((4-(羧基甲氧基)丁基)(异丙基)氨基)-2,3-二苯基吡嗪1-氧化物。
[0011]
目前国内尚未有公开高效地分析这些工艺杂质及降解杂质的方法。例如,文献肺动脉高压药物selexipag含量及有关物的测定[j],马慧丽,医药科学,2016,(30):69披露的方法,未对各杂质进行定位,不能有效分离所有杂质,无法用于药物生产的实际。
技术实现要素:
[0012]
本发明的一个目的在于提供一种快速分析司来帕格相关物质的方法,以将目标产品与工艺杂质和降解杂质充分分离、显示。
[0013]
在本发明的一个方面,本发明提供了一种利用高效液相色谱法测定司来帕格原料药中有关物质的方法。所述方法的色谱条件为:色谱柱为苯基硅烷键合硅胶柱;流动相为三氟乙酸溶液和乙腈的混合溶剂。
[0014]
由此,可以将司来帕格原料药中有关物质进行快速高效的分离,可有效控制原料药的质量。该检测方法灵敏度高、专属性强、精密度高、准确性强、操作方便,可有效控制产
品的质量。
[0015]
根据本发明的一些实施例的利用高效液相色谱法测定司来帕格原料药中有关物质的方法,还可以具有以下附加技术特征:
[0016]
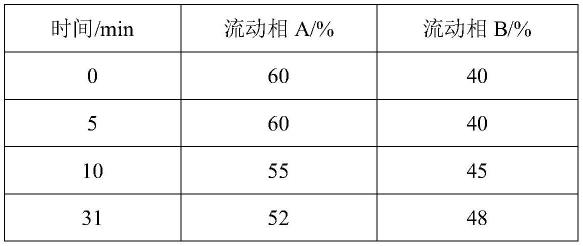
根据本发明的一种具体实施方式,梯度洗脱的条件为:
[0017]
时间/min流动相a/%流动相b/%0-575~5525-4510-3055~5245-4841-4540~2560-7546-60158561-7855~7545-25
[0018]
在本发明选定的色谱柱条件下使用该梯度程序,可以将各物质良好分离。
[0019]
根据本发明的一个较佳实施例,所述梯度洗脱的条件为:
[0020]
时间/min三氟乙酸溶液/%乙腈/%0604056040105545315248424060452575471585611585636040786040
[0021]
根据本发明的一些实施例,所述色谱柱的柱长为15cm~25cm,优选为25cm。由此,可以进一步提高分离度。
[0022]
根据本发明的一些实施例,所述三氟乙酸溶液的浓度为0.01%~0.5%,最优选为0.05%。由此,可以进一步改善峰形及空白基线干扰。
[0023]
根据本发明的实施例,本发明的分析方法进一步包括:
[0024]
采用甲醇/乙腈和水的混合液将待检测样品配制成样品溶液,将样品溶液注入高效液相色谱仪,完成司来帕格原料药中有关物质的测定。
[0025]
根据本发明的实施例,所述甲醇/乙腈和水的混合液中甲醇/乙腈和水的体积比为(50%∶50%)~(90%∶10%),优选为乙腈:水=80%∶20%。
[0026]
根据本发明的一些实施例,注入所述高效液相色谱仪的样品溶液的体积为5μl~50μl,优选为20μl。
[0027]
根据本发明的一些实施例,高效液相色谱仪的检测波长为210nm~365nm,优选为300nm。由此,可以显著提高检测灵敏度。
[0028]
根据本发明的实施例,所述流动相的流速为0.8ml/min~1.2ml/min,优选为1.0ml/min。由此,可以进一步提高分离度。
[0029]
根据本发明的一些实施例,所述色谱柱的柱温为25℃~50℃,优选为45℃。由此,
可以显著改善相邻色谱峰之间的分离度。
[0030]
根据本发明的具体实施例,测定司来帕格原料药中有关物质的方法,可以依照以下步骤实现:
[0031]
(1)取待检测样品适量,用乙腈:水(体积比为80∶20)的混合液溶解,配制成每1ml约含0.6mg/ml的样品溶液。
[0032]
(2)设置色谱柱柱温25℃~50℃,优选为45℃。流动相的流速为0.8~1.2ml/min,优选为1.0ml/min,检测波长为210nm~365nm,优选为300nm。
[0033]
(3)取(1)的样品溶液进样量5μl~50μl,优选为20μl,注入高效液相色谱仪,完成司来帕格原料药中有关物质的测定。
[0034]
本发明可将司来帕格原料药中有关物质各成分分离彻底,主峰前后杂质峰与主峰分离度良好,可操作性强,分析方法专属性强、精密度高、准确性强、操作方便,可有效控制司来帕格原料药的质量。
[0035]
本发明的附加方面和优点将在下面的描述中部分给出,部分将从下面的描述中变得明显,或通过本发明的实践了解到。
附图说明
[0036]
图1:按照实施例1条件所得空白溶液的高效液相色谱图;
[0037]
图2:按照实施例1条件所得系统适用性溶液的高效液相色谱图;
[0038]
图3:按照实施例1条件所得杂质混合溶液的高效液相色谱图;
[0039]
图4:按照实施例1条件所得加标供试品溶液的高效液相色谱图;
[0040]
图5:按照实施例2条件所得供试品溶液的高效液相色谱图;
[0041]
图6:按照实施例2条件所得对照溶液的高效液相色谱图;
[0042]
图7:按照实施例3条件所得混合溶液1的高效液相色谱图;
[0043]
图8:按照实施例4条件所得混合溶液1的高效液相色谱图;
[0044]
图9:按照实施例5条件所得混合溶液2的高效液相色谱图;
[0045]
图10:按照对比例1条件所得混合溶液2的高效液相色谱图;
[0046]
图11:按照对比例2条件所得混合溶液2的高效液相色谱图。
具体实施方式
[0047]
提供以下实施例以进一步举例说明本发明及其实施方式。在实施例中给出的具体详细内容仅是出于举例说明的目的,而不是应将其解释为限制本发明。实施例中未注明具体技术或条件的,按照本领域内的文献所描述的技术或条件或者按照产品说明书进行。所用试剂或仪器未注明生产厂商者,均为可以通过市购获得的常规产品。
[0048]
本发明实施例中所用的司来帕格原料药为申请人按上述合成路线自制。
[0049]
实施例1
[0050]
仪器:高效液相色谱仪,带二极管阵列检测器
[0051]
色谱柱:supelcosil lc-dp(4.6mm
×
250mm,5μm)
[0052]
流动相a:0.05%三氟乙酸溶液
[0053]
流动相b:乙腈
[0054]
梯度洗脱见下表:
[0055]
时间/min流动相a/%流动相b/%0604056040105545315248424060452575471585611585636040786040
[0056]
流速:1.0ml/min
[0057]
检测波长:300nm
[0058]
柱温:45℃
[0059]
进样体积:20μl
[0060]
溶剂:乙腈:水=80:20(v/v)
[0061]
运行时间:78分钟
[0062]
1、检测波长的确定
[0063]
采用紫外-可见分光光度计对司来帕格及其所含杂质1~杂质6在200~400nm之间进行了紫外光谱扫描,结果显示司来帕格和各杂质均在300nm波长下有较强吸收,故选择300nm作为检测波长。
[0064]
2、方法学专属性
[0065]
空白溶液:乙腈-水(80:20)
[0066]
取杂质1、杂质2、杂质3、杂质4、杂质5、杂质6各对照品适量,加溶剂溶解并稀释制成浓度分别约为15μg/ml、15μg/ml、15μg/ml、75μg/ml、150μg/ml、150μg/ml的杂质对照品储备液。
[0067]
取司来帕格对照品适量,加溶剂溶解并稀释制成浓度约为75μg/ml的储备液。
[0068]
系统适用性溶液:取本品约15mg,精密称定,置100ml量瓶中,加溶剂溶解并稀释至刻度,摇匀;精密量取1ml,置50ml量瓶中,精密加入杂质4储备液、杂质5储备液、杂质6储备液各2ml,加溶剂稀释至刻度,摇匀。
[0069]
杂质混合溶液:精密量取杂质1储备液2ml、杂质2储备液2ml、杂质3储备液2ml、杂质4储备液0.6ml、杂质5储备液0.1ml、杂质6储备液0.1ml,置50ml量瓶中,加溶剂溶解并稀释至刻度,摇匀。
[0070]
加标供试品溶液:取本品约30mg,精密称定,置50ml量瓶中,精密加入杂质4储备液0.6ml、杂质5储备液0.1ml、杂质6储备液0.1ml,加溶剂溶解并稀释至刻度,摇匀。
[0071]
精密量取空白溶液、系统适用性溶液、杂质混合溶液、加标供试品溶液各20μl,注入高效液相色谱仪,记录色谱图。
[0072]
结果显示:
[0073]
(1)空白溶液(见图1)对各峰检测无干扰。
[0074]
(2)系统适用性溶液(见图2)中保留时间为13.520min、15.456min、23.679min、26.977min的色谱峰依次为杂质6、杂质5、杂质4和司来帕格的色谱峰,其中司来帕格与杂质4的分离度为5.906,杂质5与杂质6的分离度为5.370,司来帕格峰的拖尾因子为1.039,理论塔板数为37268。
[0075]
(3)杂质混合溶液(见图3)中保留时间为13.588min、15.495min、17.634min、21.597min、23.799min、47.338min的色谱峰依次为杂质6、杂质5、杂质1、杂质2、杂质4和杂质3的色谱峰,其中各峰之间最小分离度为4.367(杂质2和杂质4之间),各峰之间分离度均大于1.5。
[0076]
(4)加标供试品溶液(见图4)中保留时间为13.532min、15.460min、23.665min、26.469min的色谱峰依次为杂质6、杂质5、杂质4和司来帕格的色谱峰,其中22.577min的未知杂质峰和杂质4之间分离度最小(分离度为2.023),各峰之间分离度均大于1.5。
[0077]
以上结果表明,该分析方法的专属性强。
[0078]
3、精密度试验(重复性试验)
[0079]
精密称取司来帕格原料药约15mg,置25ml量瓶中,加乙腈-水(80:20)超声使溶解并稀释至刻度,摇匀,作为供试品溶液(平行配制6份)。
[0080]
精密量取6份供试品溶液各20μl,注入高效液相色谱仪,测定已知杂质、最大未知单杂和主峰的面积归一化含量,记录色谱,结果见表1。从表1可以看出,6份供试品溶液中主峰含量结果的rsd为0.001%,杂质4和最大未知单杂的含量rsd分别为2.4%和1.1%,说明本有关物质的检测方法的重复性良好。
[0081]
表1:重复性试验结果
[0082]
序号123456平均值rsd(%)杂质4(%)0.0160.0170.0170.0170.0170.0170.0172.4最大未知单杂(%)0.0380.0390.0380.0380.0380.0380.0381.1主峰(%)99.90499.90499.90499.90499.90399.90399.900.001
[0083]
实施例2
[0084]
仪器:高效液相色谱仪,带紫外检测器
[0085]
色谱柱:supelcosil lc-dp(4.6mm
×
250mm,5μm)
[0086]
流动相a:0.05%三氟乙酸溶液
[0087]
流动相b:乙腈
[0088]
梯度洗脱见下表:
[0089]
[0090][0091]
流速:1.0ml/min
[0092]
检测波长:300nm
[0093]
柱温:45℃
[0094]
进样体积:20μl
[0095]
溶剂:乙腈:水=80:20(v/v)
[0096]
运行时间:78分钟
[0097]
实验步骤:
[0098]
供试品溶液:取本品约15mg,精密称定,置25ml量瓶中,加溶剂溶解并稀释至刻度,摇匀。
[0099]
对照溶液:精密量取供试品溶液1ml,置100ml量瓶中,加溶剂稀释至刻度,摇匀。
[0100]
精密量取供试品溶液、对照溶液各20μl,注入高效液相色谱仪,记录色谱图,具体见图5和图6。
[0101]
采用主成分自身对照法计算司来帕格原料药中各杂质的含量,结果显示各杂质均≤0.1%,总杂质≤0.5%,原料药的有关物质检测结果符合质量标准。
[0102]
实施例3
[0103]
仪器:高效液相色谱仪,带紫外检测器
[0104]
色谱柱:supelcosil lc-dp(4.6mm
×
250mm,5μm)
[0105]
流动相a:0.05%三氟乙酸溶液
[0106]
流动相b:乙腈
[0107]
洗脱条件为:
[0108]
[0109][0110]
流速:1.0ml/min
[0111]
检测波长:300nm
[0112]
柱温:35℃
[0113]
进样体积:20μl
[0114]
运行时间:70分钟
[0115]
实验步骤:
[0116]
混合溶液1配制:取杂质1、杂质2、杂质3、杂质4、杂质5、杂质6、司来帕格各对照品适量,加甲醇-水(80:20)溶解并稀释制成浓度各约为2.5μg/ml的混合溶液。
[0117]
取混合溶液1,在上述色谱条件下进行高效液相色谱分析,记录色谱图,结果见附图7。
[0118]
图7中保留时间为11.234min、13.759min、17.831min、22.837min、26.589min、32.044min、45.714min的色谱峰分别为杂质6、杂质5、杂质1、杂质2、杂质4、司来帕格、杂质3的色谱峰,司来帕格与杂质4的分离度为6.176,杂质6与杂质5的分离度为5.605,其余各峰之间的分离度均大于4.0。
[0119]
以上色谱图表明司来帕格原料药中有关物质可以达到良好的分离。
[0120]
实施例4
[0121]
仪器:高效液相色谱仪,带紫外检测器
[0122]
色谱柱:supelcosil lc-dp(4.6mm
×
250mm,5μm)
[0123]
流动相a:0.1%三氟乙酸溶液
[0124]
流动相b:乙腈
[0125]
梯度洗脱见下表:
[0126]
[0127][0128]
流速:1.0ml/min
[0129]
检测波长:300nm
[0130]
柱温:30℃
[0131]
进样体积:20μl
[0132]
运行时间:66分钟
[0133]
实验步骤:
[0134]
混合溶液1配制:取杂质1、杂质2、杂质3、杂质4、杂质5、杂质6、司来帕格各对照品适量,加甲醇-水(80:20)溶解并稀释制成浓度各约为2.5μg/ml的混合溶液。
[0135]
取混合溶液1,在上述色谱条件下进行高效液相色谱分析,记录色谱图,结果见附图8。
[0136]
图8中保留时间为22.248min、23.620min、25.218min、28.124min、28.994min、30.191min、37.829min的色谱峰分别为杂质6、杂质5、杂质1、杂质2、杂质4、司来帕格、杂质3的色谱峰,司来帕格与杂质4的分离度为3.954,杂质2与杂质4的分离度为2.865,其余各峰之间的分离度均大于2.0。
[0137]
以上色谱图表明司来帕格原料药中有关物质可以达到良好的分离。
[0138]
实施例5
[0139]
仪器:高效液相色谱仪,带紫外检测器
[0140]
色谱柱:supelcosil lc-dp(4.6mm
×
250mm,5μm)
[0141]
流动相a:0.1%三氟乙酸溶液
[0142]
流动相b:乙腈
[0143]
梯度洗脱见下表:
[0144][0145]
[0146]
流速:1.0ml/min
[0147]
检测波长:300nm
[0148]
柱温:30℃
[0149]
进样体积:20μl
[0150]
运行时间:56分钟
[0151]
实验步骤:
[0152]
混合溶液2配制:取杂质1、杂质2、杂质3、杂质4、司来帕格各对照品适量,加甲醇-水(80:20)溶解并稀释制成浓度各约为2.5μg/ml的混合溶液。
[0153]
取混合溶液2,在上述色谱条件下进行高效液相色谱分析,记录色谱图,结果见附图9。
[0154]
图9中保留时间为21.990min、23.385min、23.842min、24.638min、29.553min的色谱峰分别为杂质1、杂质2、杂质4、司来帕格、杂质3的色谱峰,其中杂质2与杂质4的分离度最小(分离度为1.953),各峰之间分离度均大于1.5。
[0155]
以上色谱图表明司来帕格原料药中有关物质可以达到良好的分离。
[0156]
对比例1
[0157]
仪器:高效液相色谱仪,带检测器
[0158]
色谱柱:wondasil c18(250mm
×
4.6mm,5μm)
[0159]
流动相a:0.1%三氟乙酸溶液
[0160]
流动相b:乙腈
[0161]
梯度洗脱见下表:
[0162]
时间/min流动相a/%流动相b/%0752557525252080452080467525567525
[0163]
流速:1.0ml/min
[0164]
检测波长:300nm
[0165]
柱温:30℃
[0166]
进样体积:20μl
[0167]
运行时间:56分钟
[0168]
实验步骤:
[0169]
混合溶液2配制:取杂质1、杂质2、杂质3、杂质4、司来帕格各对照品适量,加甲醇-水(80:20)溶解并稀释制成浓度各约为2.5μg/ml的混合溶液。
[0170]
取混合溶液2,在上述色谱条件下进行高效液相色谱分析,记录色谱图,结果见附图10。
[0171]
图10中保留时间为25.161min、25.347min、25.919min、27.963min、34.614min的色谱峰分别为杂质4、杂质2、司来帕格、杂质1、杂质3的色谱峰,其中杂质4与杂质2之间未分
开。
[0172]
以上色谱图表明在实施例5的基础上改变色谱柱类型,司来帕格原料药中有关物质不能达到良好的分离。
[0173]
对比例2
[0174]
仪器:高效液相色谱仪,带紫外检测器
[0175]
色谱柱:wondasil c18(250mm
×
4.6mm,5μm)
[0176]
流动相a:0.1%三氟乙酸溶液
[0177]
流动相b:乙腈
[0178]
梯度洗脱见下表:
[0179]
时间/min流动相a/%流动相b/%0703057030155050255050353070483070527030657030
[0180]
流速:1.0ml/min
[0181]
检测波长:300nm
[0182]
柱温:30℃
[0183]
进样体积:20μl
[0184]
运行时间:65分钟
[0185]
实验步骤:
[0186]
混合溶液2配制:取杂质1、杂质2、杂质3、杂质4、司来帕格各对照品适量,加甲醇-水(80:20)溶解并稀释制成浓度各约为2.5μg/ml的混合溶液。
[0187]
取混合溶液2,在上述色谱条件下进行高效液相色谱分析,记录色谱图,结果见附图11。
[0188]
图11中保留时间为33.060min、33.448min、35.409min、38.244min的色谱峰分别为杂质2、杂质4、司来帕格、杂质1的色谱峰,其中杂质2与杂质4之间未分开,杂质3未洗脱出来。
[0189]
以上色谱图表明在对比例1的基础上改变梯度洗脱程序,司来帕格原料药中有关物质不能达到良好的分离,也不能实现有关物质的准确测定。
[0190]
尽管上面已经示出和描述了本发明的实施例,可以理解的是,上述实施例是示例性的,不能理解为对本发明的限制,本领域的普通技术人员在本发明的范围内可以对上述实施例进行变化、修改、替换和变型。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1