一种外用跌打止痛膏的定量检测方法与流程
1.本发明涉及生物医药领域,特别是涉及一种外用跌打止痛膏的定量检测方法。
背景技术:
2.外用跌打止痛膏为中药复方制剂,处方中主要成分包括:土鳖虫、续断、防风、龙骨、马钱子、血竭、栀子、蒲公英、黄芩、金银花、虎杖、大黄、没药、儿茶、红花、紫花地丁、芙蓉、骨碎补、薄荷脑、水杨酸甲酯、冰片、樟脑共22味药,功能主治:促进骨质愈合,活血化瘀,消肿,止痛。
3.作为一种重要的中药复方制剂,外用跌打止痛膏质量检测非常重要。但是目前对外用跌打止痛膏的质量检测标准仅为载于国家食品药品监督管理总局国家药品标准ws3-b-3796-98-2016,并且该标准中仅有[性状]、[鉴别]、[挥发油]的项目检查,没有含量测定项。加之外用跌打止痛膏作为一种橡胶膏剂,其组成成分复杂,黏性大,难于分离和质量控制,特别是由于其含有大量的橡胶基质,该橡胶基质由橡胶、脂松香、凡士林、液体石蜡等物质组成,用常规的提取方法对其有效成分提取难度大,目前还不存在以其某有效成分含量对其进行质量控制的方案。
[0004]
因此,亟待提供一种能够针对外用跌打止痛膏进行定量检测的方法。
[0005]
基于此,发明人前期研究发现,君药龙骨、土鳖虫和臣药没药均无可进行有效成分检测的含量测定项,在对臣药红花的含量测定方法进行研究时发现,在对红花中的山奈素进行了含量测定的预试验时,试验结果也未能检测出山奈素。进而对处方中佐药儿茶进行含量测定进行研究发现,在测定儿茶素和表儿茶素含量时,试验结果中,成品中儿茶素的含量较高,但表儿茶素的含量很低,可能是二者在本品制备或在供试品溶液制备的过程中转化,在供试品的制备与浓度方面不能二者兼顾,且此方法的流动相不够稳定,故成品中儿茶的含量测定也无法作为能够定量检测外用跌打止痛膏的方法。
技术实现要素:
[0006]
基于此,本发明的目的之一在于提供一种外用跌打止痛膏的定量检测方法。
[0007]
包括如下技术方案:
[0008]
一种外用跌打止痛膏的定量检测方法,采用高效液相色谱法对经前处理得到的外用跌打止痛膏供试品溶液进行检测;
[0009]
所述外用跌打止痛膏的前处理为:取外用跌打止痛膏,除去盖衬、剪成条状、混匀后,得到混匀后的外用跌打止痛膏,加入乙醇提取,加热回流处理60min~180min,用乙醇补足减失的重量,摇匀,过滤,得到续滤液。
[0010]
本发明为了找到一种能够定量检测外用跌打止痛膏的方法,经过对外用跌打止痛膏中的多种成分进行检测并根据外用跌打止痛膏的剂型特点,发现在外用跌打止痛膏的供试品溶液制备中,采用合适的前处理方法对供试品进行处理,可以有效将外用跌打止痛膏中的橡胶等杂质高效洗脱分离,从而更好地保护液相色谱柱,对色谱柱耐用性好;而且,发
明人发现,当采用乙腈-水(11∶89)时,供试品溶液色谱图中栀子苷峰与杂质峰可以很好的分离,当整个检测方法,有序地配合,从而有效地实现了采用高效液相色谱法对外用跌打止痛膏中栀子苷定量检测,达到了专属性强并能够定量检测外用跌打止痛膏的方法。
附图说明
[0011]
图1为实施例3中采用i号色谱柱进行高效液相检测的结果图,其中a为栀子苷对照品,b为供试品(批号:xc006),c为缺栀子的阴性对照品。
[0012]
图2为实施例3中采用ii号色谱柱进行高效液相检测的结果图,其中a为栀子苷对照品,b为供试品(批号:xc006),c为缺栀子的阴性对照品。
[0013]
图3为实施例3中采用iii号色谱柱进行高效液相检测的结果图,其中a为栀子苷对照品,b为供试品(批号:xc006),c为缺栀子的阴性对照品。
[0014]
图4为实施例4中采用本发明实施例1的检测方法进行专属性验证检测结果,其中a为栀子苷对照品,b为供试品(批号:xc006),c为缺栀子的阴性对照品。
[0015]
图5为实施例4中采用本发明实施例1的检测方法进行线性检测结果标准曲线图。
[0016]
图6为按照实施例1的检测方法,当乙腈-水的比例为15∶85时的检测结果。
[0017]
图7为实施例2中不同洗脱溶剂的对比结果示意图,其中,1是栀子苷对照溶液,2是供试液1、3是供试液2、4是供试液3、5是供试液4。
具体实施方式
[0018]
下列实施例中未注明具体条件的实验方法,通常按照常规条件,例如sambrook等人,分子克隆:实验室手册(new york:cold spring harbor laboratory press,1989)中所述的条件,或按照制造厂商所建议的条件。实施例中所用到的各种常用化学试剂,均为市售产品。
[0019]
除非另有定义,本发明所使用的所有的技术和科学术语与属于本发明的技术领域的技术人员通常理解的含义相同。本发明的说明书中所使用的术语只是为了描述具体的实施例的目的,不用于限制本发明。本发明所使用的术语“和/或”包括一个或多个相关的所列项目的任意的和所有的组合。
[0020]
在整个说明书和权利要求书中,以下术语具有与本文明确相关的含义,除非上下文另有明确规定。在本发明中使用的短语“在一个实施方案中”不一定指代相同的实施方案,尽管其可能是。此外,在本发明中使用的短语“在另一实施方案中”不一定指代不同的实施方案,尽管其可能是。因此,如下所述,可以容易地组合本发明的各个实施方案,而不脱离本发明的范围或精神。
[0021]
本发明的技术人员在深入研究外用跌打止痛膏的检测方法时,发现紧紧围绕着栀子苷的回收率做对供试品溶液制备优化和色谱条件优化时,获得的方法检测准确度好、灵敏度高,检测结果峰型好,杂质少,能够更好地实现外用跌打止痛膏的质量控制,有利于药品安全性。
[0022]
本发明的一些实施例提供了一种外用跌打止痛膏的定量检测方法,其采用高效液相色谱法对经前处理得到的外用跌打止痛膏供试品溶液进行检测;所述外用跌打止痛膏的前处理为:取外用跌打止痛膏,除去盖衬、剪成条状、混匀后,加入提取液提取,过滤,得到续
滤液。所述提取液包括乙醇,所述提取方式为将外用跌打止痛膏进行加热回流处理30min~300min,更优选为,60min~180min。
[0023]
该检测方法专属性好,灵敏度高,能更好地实现外用跌打止痛膏的质量控制,有利于药品安全性。
[0024]
在其中一些实施例中,上述续滤液的制备步骤为制:取外用跌打止痛膏数片,除去盖衬、剪成条状、混匀,精密称定,作为供试品;加入精密称定的乙醇,经加热回流后,取出放冷,并用乙醇补足减失的重量,摇匀,用干燥滤纸滤过,即得。
[0025]
在其中一些实施例中,上述外用跌打止痛膏是指适用于促进骨质愈合,活血化瘀,消炎,消肿,止痛。用于闭合性骨折,扭挫伤等骨科病症及风湿关节炎的外用膏药,进一步地,指含有栀子苷作为药效成分的外用膏药,优选为五羊牌跌打止痛膏。
[0026]
在其中一些实施例中,使用乙醇对外用跌打止痛膏进行加热回流处理60min~180min。进一步优选为加热回流时间在120min内,此时供试品提取中栀子苷含量随着时间的延长而增加。
[0027]
在其中一些实施例中,加入的乙醇与混匀后的外用跌打止痛膏的比例为50ml:2g-3g,优选为50ml:2.g—2.6g,更优选为50ml:2.5g。
[0028]
在其中一些实施例中,上述外用跌打止痛膏供试品溶液供试品溶液的制备还包括纯化步骤,进一步地,所述纯化步骤为通过氧化铝柱富集纯化。
[0029]
在其中一些实施例中,将续滤液10ml上样于氧化铝层析柱中,用乙醚25-35ml、乙醇45-55ml、20
±
1%乙醇45-55ml依次洗脱,收集20
±
1%乙醇洗脱液,蒸干,残渣加甲醇溶液溶解,转移至5ml量瓶中并定容至刻度,摇匀,即得。
[0030]
在其中一些实施例中,上述甲醇溶液中甲醇浓度为体积分数15%-25%。进一步优选为20%。
[0031]
其中用乙醚可以去除极性小的杂质及残余橡胶,凡士林等油溶性杂质、接着用乙醇是为了洗脱掉极性大的和水溶性杂质、从而最后采用20%乙醇50ml洗脱,可以实现栀子苷完全洗脱的效果。
[0032]
在研究时发现,栀子苷极性较大,易被中性氧化铝吸附,而选用极性较大的溶剂洗脱,使用洗脱溶剂体积小,洗脱比较完全,以回收率来考察本发明的检测方法。
[0033]
在其中一些实施例中,上述供试品溶液的制备中使用的氧化铝柱为中性氧化铝柱,柱内径为2.0
±
0.2cm,填料粒径为100~200目,填料量为5~15g。选择该合适的氧化铝柱配合本发明所述的合适的洗脱方式,可以最大程度除去酸类、黄铜类、酚类、鞣质等杂质。
[0034]
在其中一些实施例中,上述高效液相色谱法的流动相为乙腈水溶液,进一步地,以乙腈-水(11∶89)为流动相。
[0035]
在其中一些实施例中,上述高效液相色谱法的色谱条件包括:
[0036]
色谱柱:以十八烷基硅烷键合硅胶为填充剂;
[0037]
检测波长:238nm;
[0038]
进样量:10μl;
[0039]
流速:1.0ml/min;
[0040]
柱温:20-30℃;
[0041]
理论塔板数按栀子苷峰计算应不低于10000。
[0042]
在其中一些实施例中,上述高效液相色谱法的色谱柱选自kromasil 100-5c18、agilent eclipse xdb-c18、hypersil ods2中的任意一种。
[0043]
本发明所述检测方法与其他方法相比,没有使用毒性试剂,试剂用量少,具备良好的实际应用推广前景。
[0044]
为了便于理解本发明,下面将对本发明进行更全面的描述。本发明可以以许多不同的形式来实现,并不限于本文所描述的实施例。相反地,提供这些实施例的目的是使对本发明公开内容的理解更加透彻全面。
[0045]
以下结合具体实施例对本发明作进一步详细的说明。
[0046]
实施例1
[0047]
仪器与试剂
[0048]
仪器:waters 2695液相色谱仪;
[0049]
试剂:乙腈为色谱纯,其它试剂均为分析纯。
[0050]
对照品:栀子苷(批号110749-200714,供含量测定用),购于中国食品药品检定研究院。
[0051]
供试品:五羊牌跌打止痛膏,批号为vc011、vc003、uc05、uc02、tc04、xc005、vc004、uc04、uc03、tc03、xc006。
[0052]
栀子苷阴性对照品:按本品处方及制法,制成缺栀子药材的阴性对照样品。取相当于供试品的量,按供试品溶液的制备方法制成阴性对照品溶液。
[0053]
色谱条件:
[0054]
色谱柱:以十八烷基硅烷键合硅胶为填充剂的色谱柱;
[0055]
流动相:乙腈-水,等度洗脱,初始比例为(11:89)。
[0056]
流速:1.0ml/min;柱温:20-30℃;进样量:10μl;检测波长为238nm;理论板数以栀子苷峰计算不低于10000。
[0057]
对照品溶液的制备:精密称取栀子苷对照品,加20%甲醇制成每1ml含50μg的溶液,即得。
[0058]
供试品溶液的制备:
[0059]
取外用跌打止痛膏10片(每片面积大于35cm2的应切成35cm2),除去盖衬,精密称定,剪成条状,混匀,取约2.5g,精密称定,精密加入乙醇50ml,称定重量,加热回流2小时,取出,放冷,再称定重量,用乙醇补足减失的重量,摇匀,滤过,精密量取续滤液10ml,加在中性氧化铝柱(100~200目,10g,内径为2.0cm)上,用乙醚30ml、乙醇50ml、20%乙醇50ml依次洗脱,收集20%乙醇洗脱液,蒸干,残渣加20%甲醇使溶解,转移至5ml量瓶中并定容至刻度,摇匀,即得。
[0060]
测定法:分别精密吸取对照品溶液与供试品溶液各10μl,注入液相色谱仪,测定,本品每100cm2含栀子以栀子苷(c
17h24o10
)计,不得少于0.70mg。
[0061]
供试品应出现与栀子苷对照品色谱保留时间一致的色谱峰;且供试品中的色谱峰面积应不小于对照品栀子苷色谱峰面积。
[0062]
发明人在实验中发现,当在乙腈-水的比例为15∶85时,供试品溶液色谱图中栀子苷峰中包含杂质峰(参见图6),而当发明人将比例调整为乙腈-水(11∶89)时,发现供试品溶液色谱图中栀子苷峰与杂质峰可以较好的分离(可参考图1-4)。
[0063]
实施例2供试品溶液制备工艺优化
[0064]
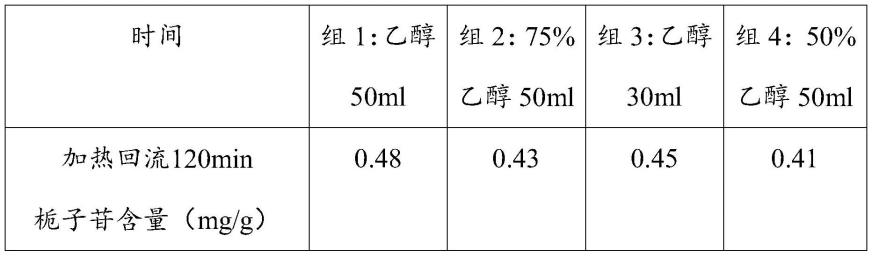
1.1提取溶剂和时间的对比
[0065]
取本品(批号:xc006),除去盖衬,剪成条状,取约2.5g,每个时间2份,精密称定,分别精密加入(组1:乙醇50ml;组2:75%的乙醇50ml;组3:乙醇30ml;组4:50%的乙醇50ml)称定重量,分别加热回流2小时,取出,放冷,再称定重量,用乙醇补足减失的重量,摇匀,滤过,精密量取续滤液10ml,加在中性氧化铝柱(100~200目,10g,内径为2.0cm)上,用乙醚30ml、乙醇50ml、20%乙醇50ml依次洗脱,收集20%乙醇洗脱液,蒸干,残渣加20%甲醇使溶解,转移至5ml量瓶中并定容至刻度,摇匀,即得。
[0066]
检测结果汇总后见下表2.1所示,
[0067]
表2.1不同提取溶剂的比较(n=2)
[0068][0069]
结果描述:用50ml乙醇作为提取溶剂,栀子苷含量最高,检测效果最佳。
[0070]
1.2提取时间的对比
[0071]
取本品(批号:xc006),除去盖衬,剪成条状,取约2.5g,每个时间2份,精密称定,分别精密加入乙醇50ml,称定重量,分别加热回流30min、1小时、2小时、3小时,取出,放冷,再称定重量,用乙醇补足减失的重量,摇匀,滤过,精密量取续滤液10ml,加在中性氧化铝柱(100~200目,10g,内径为2.0cm)上,用乙醚30ml、乙醇50ml、20%乙醇50ml依次洗脱,收集20%乙醇洗脱液,蒸干,残渣加20%甲醇使溶解,转移至5ml量瓶中并定容至刻度,摇匀,即得。
[0072]
检测结果汇总后见下表2.2所示,加热回流时间在120min内栀子苷含量随着时间的延长而增加,但加热回流180min测定的栀子苷含量与120min无显著差异,所以选择加热回流时间为120min(2小时)。
[0073]
表2.2提取时间的比较(n=2)
[0074][0075]
1.3洗脱溶剂体积的对比
[0076]
取本品(批号:xc006),除去盖衬,剪成条状,取约2.5g,精密称定,精密加入乙醇50ml,称定重量,加热回流2小时,取出,放冷,再称定重量,用乙醇补足减失的重量,摇匀,滤过。
[0077]
组1:精密量取续滤液10ml,加在中性氧化铝柱(100~200目,10g,内径为2.0cm)上,用乙醚30ml、乙醇50ml、20%乙醇50ml依次洗脱;分别收集各组最后的洗脱液,蒸干,残
渣加20%甲醇使溶解,洗脱部位转移至5ml量瓶中并定容至刻度。
[0078]
再用20%乙醇30ml洗脱,收集最后一次洗脱剂,蒸干,残渣加甲醇1ml溶解,转移至1ml容量瓶定容至刻度备用(供试液1)。
[0079]
组2:精密量取续滤液10ml,加在中性氧化铝柱(同组1),用乙醚30ml、乙醇50ml、50%乙醇50ml依次洗脱;分别收集各组最后的洗脱液,蒸干,残渣加20%甲醇使溶解,洗脱部位转移至5ml量瓶中并定容至刻度。
[0080]
再用20%乙醇30ml洗脱,收集最后一次洗脱剂,蒸干,残渣加甲醇1ml溶解,转移至1ml容量瓶定容至刻度备用(供试液2)。
[0081]
组3:精密量取续滤液10ml,加在中性氧化铝柱(同组1),用乙醚30ml、20%乙醇50ml、乙醇50ml依次洗脱;分别收集各组最后的洗脱液,蒸干,残渣加20%甲醇使溶解,洗脱部位转移至5ml量瓶中并定容至刻度。
[0082]
再用20%乙醇30ml洗脱,收集最后一次洗脱剂,蒸干,残渣加甲醇1ml溶解,转移至1ml容量瓶定容至刻度备用(供试液3)。
[0083]
组4:精密量取续滤液10ml,加在中性氧化铝柱(同组1),用乙醚30ml、乙醇50ml、20%乙醇45ml依次洗脱。分别收集各组
[0084]
最后的洗脱液,蒸干,残渣加20%甲醇使溶解,洗脱部位转移至5ml量瓶中并定容至刻度。
[0085]
再用20%乙醇30ml洗脱,收集最后一次洗脱剂,蒸干,残渣加甲醇1ml溶解,转移至1ml容量瓶定容至刻度,备用(供试液4)。
[0086]
取栀子苷(批号:110749-201718)对照品适量,加甲醇制成每1ml含1mg该成分的溶液,作为对照溶液。分别吸取对照品1ul,组1、组2、组3、组4供试液各2ul,点于同一硅胶h薄层板上,以乙酸丁酯-甲酸-水(7:2.5:2.5上层溶液为展开剂,展开,取出,晾干,紫外灯(365nm)下检视。
[0087]
结果:在相同条件下,将组1、组4的氢氧化铝柱,分别再用20%乙醇30ml洗脱,洗脱部位已检测不到栀子苷,而在在相同条件下,将组2、组3的氢氧化铝柱,分别最后再用20%乙醇30ml洗脱,洗脱部位仍检测到少量栀子苷。请见图7。
[0088]
结果表明:第三次用20%乙醇50ml过氢氧化性铝柱可以洗脱完全。
[0089]
20%乙醇45ml洗脱部位已检测不到栀子苷,表明:用20%乙醇50ml可以洗脱完全。
[0090]
实施例3
[0091]
(1)加乙醚脱脂与不脱脂的比较:
[0092]
加乙醚脱脂:取本品(批号:xc006),除去盖衬,取约2.5g,精密称定,加乙醚30ml,轻微振摇,静置后倾出上清液,反复操作3次,挥干乙醚,精密加入乙醇50ml,称定重量,加热回流2小时,取出,放冷,再称定重量,用乙醇补足减失的重量,摇匀,滤过,精密量取续滤液10ml,加在中性氧化铝柱(100~200目,10g,内径为2.0cm)上,用乙醚30ml、乙醇50ml、20%乙醇50ml依次洗脱,收集20%乙醇洗脱液,蒸干,残渣加20%甲醇使溶解,转移至5ml量瓶中并定容至刻度,摇匀,即得。
[0093]
不脱脂:取本品(批号:xc006),除去盖衬,取约2.5g,精密称定,精密加入乙醇50ml,称定重量,加热回流2小时,取出,放冷,再称定重量,用乙醇补足减失的重量,摇匀,滤过,精密量取续滤液10ml,加在中性氧化铝柱(100~200目,10g,内径为2.0cm)上,用乙醚
30ml、乙醇50ml、20%乙醇50ml依次洗脱,收集20%乙醇洗脱液,蒸干,残渣加20%甲醇使溶解,转移至5ml量瓶中并定容至刻度,摇匀,即得。
[0094]
检测结果汇总后见下表3所示,二者无显著差异,故采用不脱脂的制备方法。
[0095]
表3加乙醚脱脂与不脱脂的比较
[0096] 加乙醚脱脂(n=2)不脱脂(n=1)栀子苷含量(mg/g)0.480.48
[0097]
发明人在实验中发现:加乙醚脱脂和不加乙醚脱脂,其他步骤相同,检测结果表明,栀子苷含量相同,二者无显著差异,本实验中不进行该步骤也能取得较好效果,主要原因是后面的操作步骤中过中性氧化铝柱时先用乙醚洗脱,也可以除去脂溶性杂质。故前面的操作步骤采用不脱脂的制备方法。
[0098]
(2)不同色谱柱的比较
[0099]
取1批样品(批号:xc006)2份,按含量测定法,分别用ⅰ号:kromasil 100-5c18(200
×
4.6mm),柱号:e103072;ⅱ号:agilent eclipse xdb-c18 5μm(4.6
×
150mm);柱号:993967-902;ⅲ号:依利特hypersil ods2(5μm 4.6mm
×
150mm),柱号:e2523888,三种品牌的色谱柱测定。
[0100]
表4不同色谱柱的测定结果
[0101]
批号ⅰ柱含量ⅱ柱含量ⅲ柱含量平均值rsd%xc0060.494mg/g0.507mg/g0.510mg/g0.50mg/g1.7
[0102]
结合上表4及检测结果图1-4可知,结果三种品牌的色谱柱的分离度均达到1.5以上,理论板数达到10000以上,rsd=1.7%。
[0103]
从以上实验结果可以看出,本技术所述前处理的方法,不需要额外脱脂这步也能得到很好的供试品,且得到的供试品对于色谱柱的适应性较强。
[0104]
实施例4本发明实施例1所述检测方法的性能研究
[0105]
(1)准确度
[0106]
取同一批已知含量(批号:xc006,栀子苷含量0.49mg/g的供试品6份,每份取样量约1.25g,以当前取样含量的1:1,精密加入栀子苷对照品(0.625mg),按含量测定方法测定,计算栀子苷的回收率。结果:栀子苷平均回收率为95.4%,rsd=0.6%(见表5)。
[0107]
表5栀子苷准确度试验结果(n=6)
[0108]
[0109][0110]
(2)重复性
[0111]
取同一批供试品(批号:xc006)6份,按含量测定方法测定。结果整理统计后,如下表6所示,表明检测重复性良好。
[0112]
表6重复性试验结果(n=6)
[0113][0114]
(3)专属性
[0115]
用本发明实施例1所述方法检测外用跌打止痛膏中栀子苷含量的专属性研究。
[0116]
1、仪器与试剂
[0117]
仪器:waters 2695液相色谱仪;
[0118]
对照品:栀子苷(批号110749-200714,供含量测定用),购于中国食品药品检定研究院。
[0119]
试剂:乙腈为色谱纯,其它试剂均为分析纯。
[0120]
色谱条件:
[0121]
色谱柱:以十八烷基硅烷键合硅胶为填充剂的色谱柱;
[0122]
流动相:乙腈-水,等度洗脱,初始比例为(11:89)。
[0123]
流速:1.0ml/min;柱温:20℃-30℃;进样量:10μl;检测波长为238nm;理论板数以栀子苷峰计算不低于10000。
[0124]
2、样品制备
[0125]
对照品溶液:
[0126]
精密称取栀子苷对照品,加20%甲醇制成每1ml含50μg的溶液,即得。
[0127]
供试品溶液:
[0128]
取外用跌打止痛膏10片(每片面积大于35cm2的应切成35cm2),除去盖衬,精密称定,剪成条状,混匀,取约2.5g,精密称定,精密加入乙醇50ml,称定重量,加热回流2小时,取出,放冷,再称定重量,用乙醇补足减失的重量,摇匀,滤过,精密量取续滤液10ml,加在中性氧化铝柱(100~200目,10g,内径为2.0cm)上,用乙醚30ml、乙醇50ml、20%乙醇50ml依次洗脱,收集20%乙醇洗脱液,蒸干,残渣加20%甲醇使溶解,转移至5ml量瓶中并定容至刻度,摇匀,即得。
[0129]
精密称取缺栀子苷阴性对照品,加20%甲醇制成每1ml含50μg的溶液,即得。
[0130]
精密吸取对照品溶液、供试品溶液和阴性对照溶液各10μl,分别注入液相色谱仪,按含量测定的方法测定。结果阴性对照溶液中,在与栀子苷对照品的相应位置上无色谱峰干扰(见图4)。
[0131]
经上述方法学验证试验,结果表明所建立的试验方法在小的变动范围内对测定无影响,适用于本品栀子苷的含量测定。
[0132]
(4)线性考察
[0133]
精密吸取浓度为0.0517mg/ml的栀子苷对照品溶液各1μl、2μl、8μl、14μl、20μl、26μl、32μl,注入液相色谱仪,按含量测定的方法测定,以对照品进样量(μg)为横坐标,测得的峰面积为纵坐标,绘制标准曲线,得栀子苷的回归方5程:a=1535.8953x-1.4580,r=0.99999,结果表明,栀子苷对照品进样量在0.0517μg~1.6544μg之间与其峰面积呈良好的线性关系(见表7、图5)。
[0134]
表7线性考察结果
[0135][0136]
实施例5供试品含量限度测定
[0137]
仪器与试剂
[0138]
仪器:waters 2695液相色谱仪;
[0139]
试剂:乙腈为色谱纯,其它试剂均为分析纯。
[0140]
对照品:栀子苷(批号110749-200714,供含量测定用),购于中国食品药品检定研究院。
[0141]
供试品:五羊牌跌打止痛膏,批号为vc011、vc003、uc05、uc02、tc04、xc005、vc004、uc04、uc03、tc03、xc006。
[0142]
色谱条件:
[0143]
色谱柱:以十八烷基硅烷键合硅胶为填充剂的色谱柱;
[0144]
流动相:乙腈-水,等度洗脱,初始比例为(11:89)。
[0145]
流速:1.0ml/min;柱温:20℃-30℃;进样量:10μl;检测波长为238nm;理论板数以
栀子苷峰计算不低于10000。
[0146]
对照品溶液的制备:精密称取栀子苷对照品,加20%甲醇制成每1ml含50μg的溶液,即得。
[0147]
供试品溶液的制备:
[0148]
取外用跌打止痛膏10片(每片面积大于35cm2的应切成35cm2),除去盖衬,精密称定,剪成条状,混匀,取约2.5g,精密称定,精密加入乙醇50ml,称定重量,加热回流2小时,取出,放冷,再称定重量,用乙醇补足减失的重量,摇匀,滤过,精密量取续滤液10ml,加在中性氧化铝柱(100~200目,10g,内径为2.0cm)上,用乙醚30ml、乙醇50ml、20%乙醇50ml依次洗脱,收集20%乙醇洗脱液,蒸干,残渣加20%甲醇使溶解,转移至5ml量瓶中并定容至刻度,摇匀,即得。
[0149]
含量限度的确定:根据本品处方计算,每100cm2含栀子药材0.06433g,按《中国药典》2010年版一部“栀子”【含量测定】限度规定,栀子中栀子苷含量为不少于1.8%,以转移率100%计算成品中含量,应为每100cm2含栀子苷不少于1.15794mg,根据测定值情况,考虑到在生产、检验环节中会有损失,拟定本品每100cm2含栀子以栀子苷(c
17h24o10
)计算,不得少于0.70mg。
[0150]
取上述供试品11批,按上述含量限度测定方法测定。结果见下表8所示。
[0151]
表8供试品含量测定结果
[0152]
[0153][0154]
结合上表8检测数据可知,11批外用跌打止痛膏的样品中,每100cm2含栀子以栀子苷(c
17h24o10
)计算,均大于0.70mg,满足含量限度测定要求。
[0155]
以上所述实施例仅表达了本发明的几种实施方式,其描述较为具体和详细,但并不能因此而理解为对发明专利范围的限制。应当指出的是,对于本领域的普通技术人员来说,在不脱离本发明构思的前提下,还可以做出若干变形和改进,这些都属于本发明的保护范围。因此,本发明专利的保护范围应以所附权利要求为准。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1