药物中苯甲酸、苯甲醛、苯甲醇和氯化苄的检测方法与流程
本发明涉及一种药物中苯甲酸、苯甲醛、苯甲醇和氯化苄的检测方法。
背景技术:
1、苯甲酸苄酯是秘鲁香胶内的一种成分,存在于多种植物中,在体内迅速水解成苯甲酸和苯甲醇,苯甲醇进一步代谢成马尿酸,通过尿液排泄。
2、苯甲酸苄酯制备工艺一般为以下3种:
3、1)苯甲酸与苯甲醇直接酯化:
4、
5、2)苯甲酸甲酯与苯甲醇进行酯交换:
6、
7、3)氯化苄与苯甲酸钠酯化:
8、
9、其中路线3)为工业上常用的合成路线,在原料来源、物料成本、反应转化率、反应条件及设备要求等方面,均具有明显优势。然而,该合成路线可能有氯化苄的残留,氯化苄进一步水解为杂质苯甲酸、苯甲醛和苯甲醇。根据emea中《遗传毒性杂质限度指导原则》,氯化苄为基因毒性杂质,其能直接或间接作用于dna,从而引起基因突变。苯甲酸苄酯已收载于fda“上市药品非活性成分数据库”,口服给药:最大剂量5.6mg;肌肉注射:最大剂量371mg。以ttc(毒理学关注阈值)为1.5μg/天进行评估,要求苯甲酸苄酯中任一单一基因毒性杂质限度为50ppm。氯化苄为2a类致癌物、1类基因毒性杂质,ich m7附录中以该化合物举例计算其可接受摄入量ai值为40.6μg/天。该浓度限度极低,且基因毒性杂质反应活性高、稳定性差,极易水解为苯甲醇、进一步氧化为苯甲酸和苯甲醛。因此药物中此类痕量杂质的分析极具挑战性,要求分析方法具有较高的灵敏度和专属性。
10、氯化物(如氯化苄)的测定通常采用中国药典2020版通则0801氯化物测定法,限度控制为氯化物不得超过0.035%;苯甲醛和苯甲酸的测定采用国外药典中醛含量和酸含量等检测项目,如采用特征理化反应,利用醛的还原性,在usp中醛的测定原理可以为醛与盐酸羟胺反应成肟,用氢氧化钠滴定液滴定;酸的测定可以基于酸碱中和原理。换算后限度控制以苯甲酸计不得超过0.07%、以苯甲醛计不得超过0.05%、氯化苄不得超过0.035%。然而,上述方法均缺乏专属性,判断受主观因素影响,且属于限度检测法,无法定量各杂质含量,例如:氯化物测定以是否产生沉淀为判断依据;醛含量测定以颜色从黄色变成浅绿色为判断依据,且存在黄绿色等中间色干扰;酸含量测定通过消耗氢氧化钠滴定液体积计算。
11、苯甲酸、苯甲醛和氯化苄均含有苯环特征官能团,均有紫外吸收,如usp药典中苯扎氯铵即用液相色谱法对苯甲醛和氯化苄的液相色谱法。该方法采用梯度洗脱方式,用十八烷基硅烷键合硅胶为填充剂(推荐symmetry c18柱或ymc ods-hypersil柱,5μm,4.6mm*25cm),以钠盐溶液(1-己烷磺酸钠1.09g与磷酸二氢钠6.9g加水稀释至1000ml,摇匀,用磷酸调节ph至3.5)为流动相a,甲醇为流动相b;柱温为40℃;流速为1.0ml/min。按下表进行梯度洗脱,苯甲醇及氯化苄的检测波长为210nm,苯甲醛的检测波长为257nm。
12、
13、
14、上述方法操作较为复杂,采用离子对试剂为流动相a,且检测时间长达65min,苯甲醛出峰在梯度洗脱波峰上,苯甲醇和氯化苄检测波长为210nm,受离子对影响,基线不稳。
15、因此,需要设计一种高效检测苯甲酸、苯甲醛、苯甲醇和氯化苄的检测分析方法。
技术实现思路
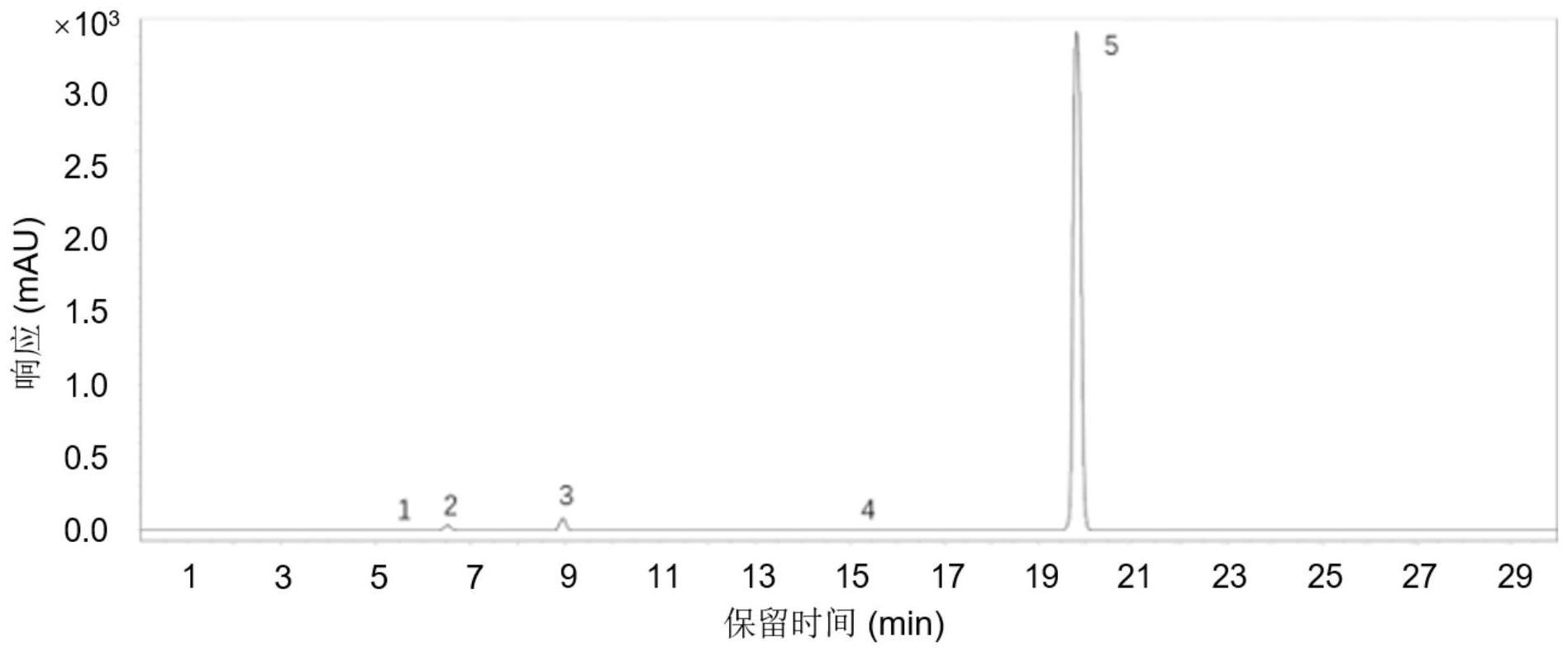
1、本发明实际所要解决的技术问题是为了克服现有技术中缺乏高效检测苯甲酸、苯甲醛、苯甲醇和氯化苄的定量分析方法的缺陷,提供了一种药物中苯甲酸、苯甲醛、苯甲醇和氯化苄的检测方法。本发明的检测方法灵敏度高、分离度好、专属性强,能够有效、快捷地检测苯甲酸、苯甲醛、苯甲醇和氯化苄,且具有良好的重复性,易于推广。
2、本发明通过以下技术方案解决上述技术问题。
3、在对苯甲酸、苯甲醛、苯甲醇和氯化苄进行检测时,由于苯甲酸极性大且易电离,出现分子形式与离子形式共存,使得苯甲酸和苯甲醇出峰接近,不易分离,甚至难以分离基线。发明人创造性地发现,在流动相a和流动相b中加入特定浓度的磷酸,配合其他技术特征,不仅可以有效分离苯甲酸和苯甲醇,还能够同时有效分析苯甲醛和氯化苄。
4、本发明提供了一种药物中苯甲酸、苯甲醛、苯甲醇和氯化苄的检测方法,其包括如下步骤:通过高效液相色谱法检测供试品溶液,根据外标法或标准曲线法计算含量即可;
5、其中,所述供试品溶液为含有待测药物的溶液;
6、所述高效液相色谱法中,流动相a为磷酸-水,流动相b为磷酸-乙腈;所述流动相a和所述流动相b中磷酸含量独立地为0.01~0.1%,%指磷酸占水或乙腈的体积比。
7、本发明中,所述高效液相色谱法中,若采用盐类试剂(如磷酸盐或乙酸铵)代替流动相a或流动相b中的磷酸,或将流动相b改为其他有机试剂(如甲醇),会导致出峰太晚、产生基线噪声、干扰检测等问题。同时,若磷酸含量太低,则苯甲酸出峰峰形异常,若磷酸含量过高,将影响色谱柱寿命。
8、本发明中,所述高效液相色谱法中,所述流动相a中磷酸含量优选为0.02~0.1%,例如0.05%。
9、本发明中,所述高效液相色谱法中,所述流动相b中磷酸含量优选为0.02~0.1%,例如0.05%。
10、本发明中,所述高效液相色谱法中,优选地,采用梯度洗脱方式,所述流动相a和所述流动相b的梯度洗脱条件如下所述:
11、 时间/min 流动相a/% 流动相b/% 0 90~50 10~50 15 50~10 50~90 30 50~90 50~10
12、上述百分比为流动相a或流动相b的体积与“流动相a和流动相b”总体积之比。
13、上表中的梯度洗脱程序如下:初始状态流动相a的体积百分比为90~50%,流动相b的体积百分比为10~50%;在0~15min的时间段内,流动相a的体积百分比由90~50%线性降低至50~10%,流动相b的体积百分比由10~50%线性上升至50~90%;在15~30min的时间段内,流动相a的体积百分比由50~10%线性上升至50~90%,流动相b的体积百分比由50~90%线性降低至50~10%。所述流动相a和所述流动相b的体积百分比以所述流动相总体积为基准。
14、更优选地,所述流动相a和所述流动相b的梯度洗脱条件如下所述:
15、 时间/min 流动相a/% 流动相b/% 0 80~60 20~40 15 40~20 60~80 30 60~80 40~20
16、上表中的梯度洗脱程序如下:初始状态流动相a的体积百分比为80~60%,流动相b的体积百分比为20~40%;在0~15min的时间段内,流动相a的体积百分比由80~60%线性降低至40~20%,流动相b的体积百分比由20~40%线性上升至60~80%;在15~30min的时间段内,流动相a的体积百分比由40~20%线性上升至60~80%,流动相b的体积百分比由60~80%线性降低至40~20%。所述流动相a和所述流动相b的体积百分比以所述流动相总体积为基准。
17、进一步优选地,所述流动相a和所述流动相b的梯度洗脱条件如下所述:
18、 时间/min 流动相a/% 流动相b/% 0 70 30 15 30 70 30 70 30
19、上表中的梯度洗脱程序如下:初始状态流动相a的体积百分比为70%,流动相b的体积百分比为30%;在0~15min的时间段内,流动相a的体积百分比由70%线性降低至30%,流动相b的体积百分比由30%线性上升至70%;在15~30min的时间段内,流动相a的体积百分比由30%线性上升至70%,流动相b的体积百分比由70%线性降低至30%。所述流动相a和所述流动相b的体积百分比以所述流动相总体积为基准。
20、本发明中,所述高效液相色谱法中,进样量可为5~20μl,例如20μl。
21、本发明中,所述高效液相色谱法中,流速可为0.8~1.2ml/min,例如1.0ml/min。
22、本发明中,所述高效液相色谱法中,柱温可为25~35℃,例如30℃。
23、本发明中,所述高效液相色谱法中,检测波长可为220~257nm,例如220nm、225nm、238nm、254nm或257nm。
24、本发明中,所述高效液相色谱法中,色谱柱中填充剂的种类可为十八烷基键合硅胶。
25、本发明中,所述高效液相色谱法中,色谱柱中填充剂的颗粒度可为本领域常规,优选为3~5μm,例如5μm。
26、本发明中,所述高效液相色谱法中,色谱柱柱长可为本领域常规,优选为15~25cm,例如25cm。
27、本发明中,所述高效液相色谱法中,色谱柱内径可为本领域常规,优选为3~6mm,例如4.6mm。
28、本发明中,所述高效液相色谱法中,色谱柱可购自安捷伦公司或菲罗门公司。
29、本发明中,所述待测药物可为本领域常规的以氯化苄为原料制得的药物,例如苯甲酸苄酯、苯甲酸钠或苯扎氯铵。
30、本发明中,所述供试品溶液的制备方法可为本领域常规,优选地,将所述待测药物溶解于稀释剂中制得。其中,所述稀释剂可为磷酸-水-乙腈。
31、所述稀释剂中磷酸、水和乙腈的配比可根据药物溶解性进行调整。所述磷酸和水的体积比优选为0.05:1。所述水和乙腈的体积比优选为1:(1~10),例如1:1。
32、本发明中,所述供试品溶液的浓度可为1~5mg/ml,例如1mg/ml、2mg/ml或5mg/ml。若供试品溶液的浓度过高,则对色谱柱有较高要求,易出现过载和色谱峰分叉的问题;同时,当药物为苯甲酸苄酯时,由于苯甲酸苄酯不溶于水,而稀释剂中含水,易产生溶剂效应,不利于高浓度的苯甲酸苄酯的溶解性,苯甲酸苄酯易析出。若供试品溶液的浓度过低,易造成在同一波长上实现物质分析的响应较低,使得待测药物无响应检测不出。
33、本发明中,所述外标法公式为:cx=cr*ax/ar,cx为供试品溶液中苯甲酸、苯甲醛、苯甲醇或氯化苄的浓度(mg/ml);cr为混合对照品溶液中苯甲酸、苯甲醛、苯甲醇或氯化苄的浓度(mg/ml);ax为供试品溶液中苯甲酸、苯甲醛、苯甲醇或氯化苄的峰面积;ar为混合对照品溶液中苯甲酸、苯甲醛、苯甲醇或氯化苄的峰面积。
34、优选地,采用外标法进行计算时,供试品溶液中待测杂质与混合对照品溶液中的待测杂质是一一对应的。例如,当待测杂质为苯甲酸时,cx为供试品溶液中的苯甲酸的浓度(mg/ml),cr为混合对照品溶液中苯甲酸的浓度(mg/ml);ax为供试品溶液中苯甲酸的峰面积;ar为混合对照品溶液中苯甲酸的峰面积。
35、其中,所述混合对照品溶液可通过本领域常规方法制得,优选地,将苯甲酸对照品、苯甲醛对照品、苯甲醇对照品和氯化苄对照品分别稀释为特定浓度的苯甲酸储备溶液、苯甲醛储备溶液、苯甲醇储备溶液和氯化苄储备溶液,将其混合并稀释即可。
36、所述稀释操作可采用制备供试品溶液的稀释剂。
37、所述特定浓度优选为2~4mg/ml。
38、所述混合对照品溶液中,苯甲酸的浓度可为2ng/ml~2μg/ml,例如1μg/ml。
39、所述混合对照品溶液中,苯甲醛的浓度可为2ng/ml~2μg/ml,例如1μg/ml。
40、所述混合对照品溶液中,苯甲醇的浓度可为2~20μg/ml,例如10μg/ml。
41、所述混合对照品溶液中,氯化苄的浓度可为2~20μg/ml,例如1μg/ml。
42、本发明中,较佳地,所述供试品溶液中苯甲酸、苯甲醛、苯甲醇和氯化苄的浓度cx通过下述方法获得:
43、将所述供试品溶液按所述高效液相色谱法进行检测,得到供试品溶液中苯甲酸、苯甲醛、苯甲醇和氯化苄的峰面积ax;将所述混合对照品溶液按所述高效液相色谱法进行检测,得到混合对照品溶液中苯甲酸、苯甲醛、苯甲醇和氯化苄的峰面积ar,所述混合对照品溶液中苯甲酸、苯甲醛、苯甲醇和氯化苄的浓度cr是已知的;
44、根据外标法公式cx=cr*ax/ar,分别计算得到供试品溶液中苯甲酸、苯甲醛、苯甲醇和氯化苄的浓度cx。
45、本发明中,所述标准曲线法可通过下述步骤制得:将浓度为混合对照品溶液50%、80%、100%、120%和150%的溶液进样,进行所述高效液相色谱法检测,根据测得的峰面积和相应溶液的浓度,得到回归方程,获得标准曲线;所述标准曲线中,优选以所述峰面积为纵坐标,相应溶液的浓度为横坐标。
46、本发明中,优选地,所述标准曲线法中苯甲酸的线性方程为y=81.559x-0.4419,r2=0.9995。
47、本发明中,优选地,所述标准曲线法中苯甲醛的线性方程为y=105.83x-0.1088,r2=0.9998。
48、本发明中,所述苯甲酸的定量限可为2.450ng/ml。
49、本发明中,所述苯甲醛的定量限可为2.770ng/ml。
50、本发明中,所述苯甲醇的定量限可为2.489μg/ml。
51、本发明中,所述氯化苄的定量限可为0.113μg/ml。
52、本发明中,所述苯甲酸的浓度检测范围可为0.00245~1.4685μg/ml。
53、本发明中,所述苯甲醛的浓度检测范围可为0.00277~1.6605μg/ml。
54、本发明中,所述苯甲醇的浓度检测范围可为2.4890~25μg/ml。
55、本发明中,所述氯化苄的浓度检测范围可为0.1130~2.5μg/ml。
56、在符合本领域常识的基础上,上述各优选条件,可任意组合,即得本发明各较佳实例。
57、本发明所用试剂和原料均市售可得。
58、本发明的积极进步效果在于:
59、1、本发明的检测方法可同时检测苯甲酸、苯甲醛、苯甲醇和氯化苄。该方法专属性强;分离度好;灵敏度高,苯甲酸、苯甲醛、苯甲醇和氯化苄的定量限分别可以实现2.450ng/ml、2.770ng/ml、2.489μg/ml和0.113μg/ml;重复性好,不同实验人员、不同时间运用不同仪器对同批次供试品溶液进行检测,各组分含量的rsd值低于3%;耐用性好;准确度高,苯甲酸、苯甲醛、苯甲醇和氯化苄的加标回收率均在90~110%范围内;线性范围良好。
60、2、本发明可以在30分钟内实现对苯甲酸、苯甲醛、苯甲醇和氯化苄的定量测定,减少药物副作用,提高产品质量;且操作简便,对色谱柱及仪器要求低,既适用于实验室的检测,更适用于企业大规模生产的产品质量控制,对于产品中控等能较快得到检测结果,易于推广。
- 还没有人留言评论。精彩留言会获得点赞!