烟酸原料药中有关物质的检测方法与流程
本发明属于原料药检测,具体涉及一种烟酸原料药中有关物质的检测方法。
背景技术:
1、烟酸(nicotinic acid)属于维生素b族,用于预防和治疗烟酸缺乏症,治疗糙皮症。
2、烟酸的主流合成工艺中,采用以3-甲基吡啶作为起始物料,3-甲基吡啶经氨氧化生成3-氰基吡啶,3-氰基吡啶碱性条件下水解生成烟酰胺,烟酰胺水解生成烟酸,该反应工艺的中间体为3-氰基吡啶和烟酰胺。
3、通常在对3-甲基吡啶的提取纯化工艺中,根据物质的沸点差异采用蒸馏的方式进行纯化,而3-甲基吡啶物料中存在各种同分异构体杂质,如2-甲基吡啶及4-甲基吡啶,均与3-甲基吡啶非常相近,难以分离。
4、因此,在烟酸的合成工艺中,反应步骤多,副反应复杂,应对每一步的中间产物进行检测及控制,才能更好的把控产品的质量。
5、针对烟酸中烟酰胺的检测方法已有公开,但是,同时测定中间体3-氰基吡啶、烟酸的异构体2-吡啶甲酸、起始物料3-甲基吡啶及其同分异构体2-甲基吡啶及4-甲基吡啶杂质的方法未见报道,2020年版中国药典中规定3-氰基吡啶不得过0.25%,其他杂质未控制;药典中3-氰基吡啶的检测方法使用了薄层色谱法,该方法专属性和准确性均不佳;对于药典中未明确控制的其他杂质的控制,适用国际人用药品注册技术协调会制定的ich指导原则。
6、在烟酸有关物质的检测中,2-甲基吡啶、3-甲基吡啶、4-甲基吡啶互为同分异构体,烟酸与2-吡啶甲酸互为同分异构体,因同分异构体之间具有相似的结构和极性,在液相色谱条件下选择性相近,这些同分异构体常常难以实现分离。
7、并且,主成分烟酸及各杂质分子量小,极性大,在反向高效液相色谱中保留能力差,通常在高效液相色谱图中在2分钟以内出峰,从而难以进行分离,方法开发难度较大。因此,对烟酸原料药进行质量控制,尚有较大的探索空间。
技术实现思路
1、本发明的目的在于提供一种烟酸原料药中有关物质的检测方法,通过该方法,2-甲基吡啶、3-甲基吡啶、4-甲基吡啶、2-吡啶甲酸及3-氰基吡啶与烟酸的色谱峰分离度均不小于1.5,线性关系良好,实现对两组同分异构体的分离控制,对烟酸原料药进行质量控制。
2、为了达到上述目的,本发明提供如下技术方案:
3、烟酸原料药中有关物质的检测方法,包括以下步骤:
4、对烟酸原料药待测溶液进行高效液相色谱法检测,采用十八烷基键合硅胶色谱柱,以ph=2.0~4.0的2~10mmol/l辛烷磺酸钠溶液为流动相a,甲醇为流动相b进行梯度洗脱;检测波长:260~280nm;进样量:10~50μl;流速:0.4~0.6ml/min;色谱柱温度:25~40℃。
5、进一步,所述有关物质包括2-甲基吡啶、3-甲基吡啶、4-甲基吡啶、2-吡啶甲酸和/或3-氰基吡啶。
6、又,所述梯度洗脱过程为:以体积百分数为90%~95%的流动相a、5~10%的流动相b为起始流动相进行等度洗脱,烟酸被洗脱结束后,以95~80%的流动相a、5~20%的流动相b洗脱3~15分钟,不再有色谱峰被洗脱出后,以不低于50%的流动相b洗脱至少3分钟后,恢复起始流动相进行平衡,平衡时间不小于5分钟。
7、优选地,所述梯度洗脱过程为:以体积百分数为95%的流动相a、5%的流动相b为起始流动相进行等度洗脱,烟酸峰洗脱结束后,以体积百分数为80%的流动相a、20%的流动相b洗脱,不再有色谱峰被洗脱出后,以不低于50%的流动相b洗脱至少5分钟后,恢复至起始流动相进行平衡。
8、又,所述起始流动相的等度洗脱时间为3~10分钟,所述有关物质还包括4-吡啶甲酸和/或烟酰胺。
9、优选地,所述色谱柱选自waters x-bridge系列或thermo hypersil bds 18系列。
10、又,所述色谱柱填料粒径≤5μm,长度≥5cm,内径≤4.6mm。
11、进一步,所述流动相a中,用磷酸调节ph值。
12、优选地,所述烟酸原料药待测溶液中烟酸的浓度为0.2~0.5mg/ml。
13、又,所述流动相a中,辛烷磺酸钠溶液的浓度为2.3~5mmol/l,用磷酸调节ph值至2.5,色谱柱温度:30℃;进样量:20μl,流速:0.5ml/min。
14、本发明的待测对象中,2-甲基吡啶、3-甲基吡啶、4-甲基吡啶互为同分异构体,烟酸和2-吡啶甲酸互为同分异构体,两组物质在反相色谱中没有明显可区分的特点,并且,待分离的各组分均为大极性小分子化合物,保留能力弱,在保留能力很弱的前提下,各待测组分的分离空间更为有限。因此,在一般液相色谱体系中,这些同分异构体通常难以被分离。
15、本发明中,以2~10mmol/l辛烷磺酸钠溶液作为流动相a,辛烷磺酸钠作为离子对试剂能够存在于液相色谱体系中,与色谱柱填料进行作用,增强待分离物质的保留能力,若流动相中辛烷磺酸钠含量过低,会影响2-吡啶甲酸与3-氰基吡啶的分离度,导致两者难以分离。
16、在反相色谱体系中,水相为固定相,有机溶剂为洗脱剂,流动相中水比例越高,待测组分越不溶液被洗脱,保留能力越强,然而硅胶溶于水,普通c18柱流动相中通常水比例不超过90%,本发明采用亲水的强保留色谱体系,可以耐高水比例流动相,流动相中水的比例可高达95%,使得烟酸及其同分异构体2-吡啶甲酸之间能够进行分离,另一组同分异构体中3-甲基吡啶峰与4-甲基吡啶出峰完全重合,因此做合并控制,4-甲基吡啶峰与其他待测组分均可分离。
17、在高效液相色谱检测中,进样量越大时色谱峰面积越大,但色谱峰面积过大会使检测器过载,无法准确测量;而色谱峰面积过小时,误差对检测结果的影响较大,本发明中采用10~50μl的进样量,以保证目标色谱峰面积处于合适范围,减少峰面积对分离效果的影响,提升检测结果的准确性。
18、与现有技术相比,本发明具有如下有益效果:
19、本发明中,对烟酸原料中的2-甲基吡啶、3-甲基吡啶、4-甲基吡啶、2-吡啶甲酸、3-腈基吡啶进行定量检测,使用同一色谱条件对两组同分异构体进行了分离,并且能够对各组分的含量进行有效的定量分析,具有高效性和经济性。
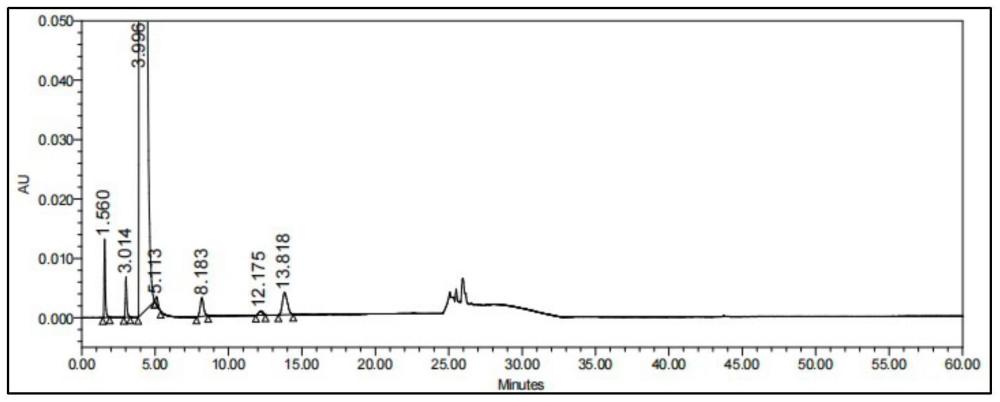
20、本发明中,各杂质之间分离度良好,各峰与烟酸峰的分离度不低于1.5,3-甲基吡啶和4-甲基吡啶出峰一致,可进行合并控制,其余色谱峰之间最小分离度为2.8,目标色谱峰峰形尖锐,对称性良好,结果可信度高,为烟酸原料药的质量检测提供了可靠的检验基础。
技术特征:
1.烟酸原料药中有关物质的检测方法,包括以下步骤:
2.根据权利要求1所述烟酸原料药中有关物质的检测方法,其特征在于,所述有关物质包括2-甲基吡啶、3-甲基吡啶、4-甲基吡啶、2-吡啶甲酸和/或3-氰基吡啶。
3.根据权利要求1所述烟酸原料药中有关物质的检测方法,其特征在于,所述梯度洗脱过程为:以体积百分数为90%~95%的流动相a、5~10%的流动相b为起始流动相进行等度洗脱,烟酸被洗脱结束后,以95~80%的流动相a、5~20%的流动相b洗脱3~15分钟,不再有色谱峰被洗脱出后,以不低于50%的流动相b洗脱至少3分钟后,恢复起始流动相进行平衡,平衡时间不小于5分钟。
4.根据权利要求3所述烟酸原料药中有关物质的检测方法,其特征在于,所述梯度洗脱过程为:以体积百分数为95%的流动相a、5%的流动相b为起始流动相进行等度洗脱,烟酸洗脱结束后,以体积百分数为80%的流动相a、20%的流动相b洗脱,不再有色谱峰被洗脱出后,以不低于50%的流动相b洗脱至少5分钟后,恢复至起始流动相进行平衡。
5.根据权利要求3所述烟酸原料药中有关物质的检测方法,其特征在于,所述起始流动相的等度洗脱时间为3~10分钟,所述有关物质还包括4-吡啶甲酸和/或烟酰胺。
6.根据权利要求1所述烟酸原料药中有关物质的检测方法,其特征在于,所述色谱柱选自waters x-bridge系列或thermo hypersil bds18系列。
7.根据权利要求6所述烟酸原料药中有关物质的检测方法,其特征在于,所述色谱柱填料粒径≤5μm,长度≥5cm,内径≤4.6mm。
8.根据权利要求1所述烟酸原料药中有关物质的检测方法,其特征在于,所述流动相a中,用磷酸调节ph值。
9.根据权利要求1所述烟酸原料药中有关物质的检测方法,其特征在于,所述烟酸原料药待测溶液中烟酸的浓度为0.2~0.5mg/ml。
10.根据权利要求1所述烟酸原料药中有关物质的检测方法,其特征在于,所述流动相a中,辛烷磺酸钠溶液的浓度为2.3~5mmol/l,用磷酸调节ph值至2.5,色谱柱温度:30℃;进样量:20μl,流速:0.5ml/min。
技术总结
烟酸原料药中有关物质的检测方法,所述有关物质包括2‑甲基吡啶、3‑甲基吡啶、4‑甲基吡啶、2‑吡啶甲酸和/或3‑氰基吡啶,采用十八烷基键合硅胶色谱柱,以pH=2.0~4.0的2~10mmol/L辛烷磺酸钠溶液为流动相A、甲醇为流动相B进行梯度洗脱;检测波长:260~280nm;进样量:10~50μl;流速:0.4~0.6ml/min;色谱柱温度:25~40℃,在同一色谱条件下对两组同分异构体实现了分离,并且能够对各组分的含量进行有效的定量分析,结果可信度高,具有高效性和经济性,为烟酸原料药的质量检测提供了可靠的检验基础。
技术研发人员:高衎,杨易可,周洁,赵帅旗,李丽霞
受保护的技术使用者:上海旭东海普药业有限公司
技术研发日:
技术公布日:2024/2/25
- 还没有人留言评论。精彩留言会获得点赞!